- Click on
to access your saved filter sets. If you enable the
 option, clicking on Filters will give you access to a preview of the redesigned dynamic filters, including AND/OR logic.
option, clicking on Filters will give you access to a preview of the redesigned dynamic filters, including AND/OR logic.
- Click on the Spreadsheet icon
to see the list of variants selected to be exported in a spreadsheet. To export the selected variant list, click on the above icon and select
and the exported spreadsheet will be downloaded automatically from your browser.
- Click on the Report generation icon
to see the list of variants selected to be included in the Report. You can download a Report of the selected variants in PDF format. To do so, click on the above icon, then select
and you will be directed to the following screen.
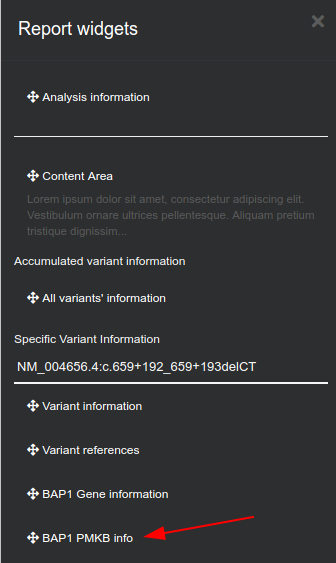
By clicking on the
icon, a Report widgets menu will be shown to customize the report.
You can drag and drop the information you prefer to include in the report. At the last option you can find PMKB information for the gene that includes the variant. The options provided here are the same as described in "Analysis actions" options.
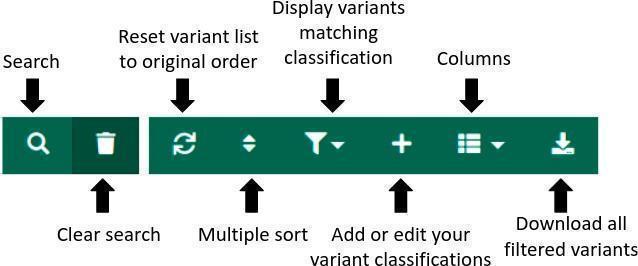
Green menu
 Search: You can search through your results by querying according to the VarSome search format. The query can include any of the following:
Search: You can search through your results by querying according to the VarSome search format. The query can include any of the following:
- gene: e.g. PIK3CA,
- chromosome: e.g. chr3 or 3
- chromosome position: e.g. chr3:178947865, chr3-178947865, chr3 178947865 or 3 178947865.
- genomic range: e.g. chr3:178936091:178942431, chr3-178936091-178942431, chr3 178936091 178942431 or 3 178936091 178942431.
- variant (DNA): e.g. chr3:178936091 G⇒A, chr3:178936091-G-A, chr3-178936091-G-A, chr3 178936091 G A, 3:178936091 G⇒A, 3:178936091-G-A…
- variant (HGVS): e.g. NM_004448.4:c.1947-3C>A
- variant (protein): e.g. BRAF:V600E or BRAF V600E.
- rsIDs ("rs" followed by a number)
- COSMIC IDs
This will filter the table and show only the results for that query.
 Clear search: This will empty the search box and show all variants again.
Clear search: This will empty the search box and show all variants again.  Reset variant list to original order: Clicking on this icon resets the sorting order of the columns to the default (the variants will be ordered by Class).
Reset variant list to original order: Clicking on this icon resets the sorting order of the columns to the default (the variants will be ordered by Class).  Multiple sorting: The list of variants can be sorted by multiple columns. A pop-up window will appear and multiple columns of interest can be selected in order to sort the variants in ascending or descending order.
Multiple sorting: The list of variants can be sorted by multiple columns. A pop-up window will appear and multiple columns of interest can be selected in order to sort the variants in ascending or descending order.
Note: Multiple-column sorting will return informative results as long as the first column, which is selected to sort the variants, has numeric values (Frequency, number of samples, Phenotypes etc). For example, the user should not sort first by “AMP Tier” or “ACMG Class” and then sort by other values like allelic balance, frequency, etc. However “ACMG Class” and “AMP Tier” can be used as second or later in the order of columns to sort by.
 Display variants matching classification: Filters for custom variant classifications.
Display variants matching classification: Filters for custom variant classifications.Add or edit your variant classifications: Open the Custom Tag creation menu. Custom tags allow you to classify variants using user-defined tags.
Columns: Remove or add columns to the table. This functionality can be used to remove columns that are not relevant for the analysis.
Download all filtered variants from the table below (max 50000) in Excel format: Download the list of variants (max. 50000) that pass any currently applied filters in Excel format. The Excel file also contains information about the filters used to obtain the exported table.
⚠️ If you need to download a list of more than 50000 variants, you can use the VarSome Clinical API. Alternatively, you can either download the VCF without annotations or filter the variants using our dynamic filters and download the filtered set.
Blue menu
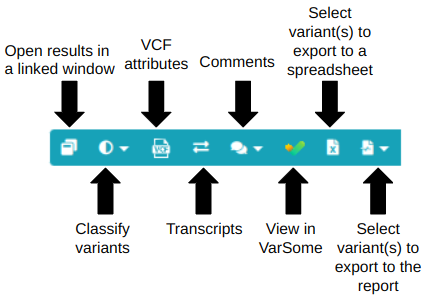
Open results in a linked window: This functionality allows you to utilize multiple screens by generating "linked" sub-windows that contain the results of an analysis. (please find detailed information in the following article: "How can I inspect my analysis results on more than one monitors?".
 Classify variants: add your own classification to a variant.
Classify variants: add your own classification to a variant. VCF attributes: pop-up window describing the quality details for each software tool used to identify the variants. For more information see the document "VCF attributes explained".
VCF attributes: pop-up window describing the quality details for each software tool used to identify the variants. For more information see the document "VCF attributes explained". Transcripts: pop-up window with all the RefSeq transcripts containing the variant. It also shows the location of the variant (intron/exon, amino acid position), its HGVS notation, and genomic function (intronic, exonic, splicing, UTR ...). Canonical transcripts are shown in red.
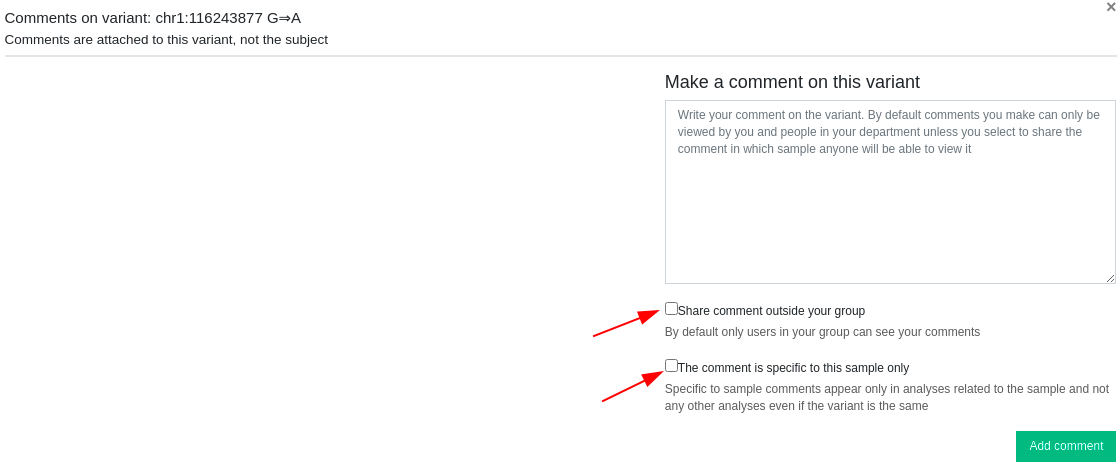
Transcripts: pop-up window with all the RefSeq transcripts containing the variant. It also shows the location of the variant (intron/exon, amino acid position), its HGVS notation, and genomic function (intronic, exonic, splicing, UTR ...). Canonical transcripts are shown in red. Comments: It is possible to attach a short comment to a selected variant (long comments will not be added and will return an error message). These comments will be linked to the variant or the gene and will be displayed in other analyses if the same variant is found. Variants with comments will have an
Comments: It is possible to attach a short comment to a selected variant (long comments will not be added and will return an error message). These comments will be linked to the variant or the gene and will be displayed in other analyses if the same variant is found. Variants with comments will have an icon in the Variant or Gene column. Comments are shared only within your group unless you decide to make your comments public by selecting the “Share comment outside your group” option. You can also select the “The comment is specific to this sample only” option and the comment will be available only to this specific sample analysis. If however, the variant is present in other analyses, the sample-specific comment will not be shown.
 View in VarSome: link to our free knowledge base and database aggregator, VarSome.
View in VarSome: link to our free knowledge base and database aggregator, VarSome.Select variant(s) to export to a spreadsheet: Clicking on this box selects the variant to export to a spreadsheet in Excel format.
Select variant(s) to export to the report: Clicking this box will allow the selection of the variant as either a Primary or Secondary finding to be included in the report.
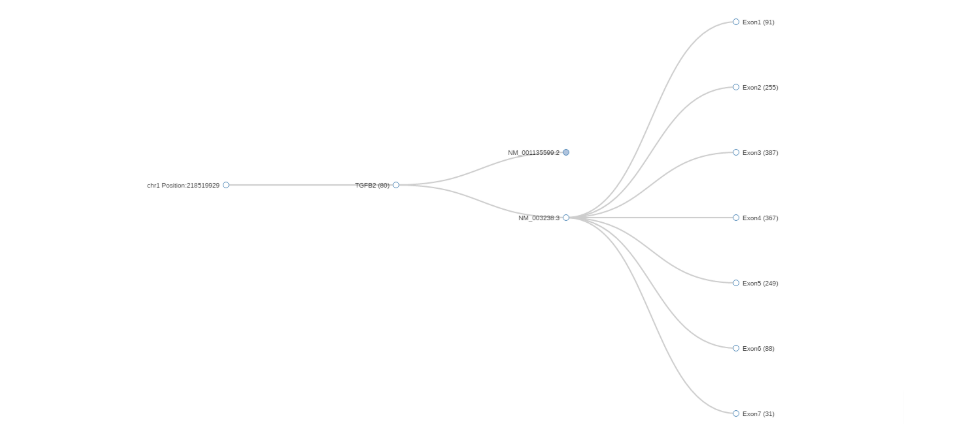
Gene coverage: a pop-up window showing the average coverage for the selected gene and its different transcripts. Clicking on the nodes will expand or collapse the tree.

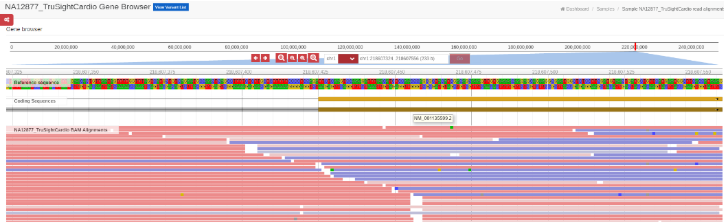
Also, by clicking on one of the Exons, a new tab will open with a JBrowse (jbrowse.org) window showing the alignment details from the analysis’ bam files. JBrowse is a software tool installed on our secured servers.

Read Alignment on JBrowse: Opens a new tab with a JBrowse representation of the BAM files.
Read Alignment on IGV: Opens a new tab with an IGV representation of the BAM files.