A VCF (Variant Calling Format) is a text file format that stores information about genetic variation. Each variant shown in the variant table corresponds to one line in the VCF file. Besides the variant position, reference and alternative alleles, each line contains additional information (attributes) such as different quality measures that can be accessed from VarSome Clinical. To see these, you need to select the variant in the table, then you can click on the VCF icon to display the VCF attributes as shown in the picture below.

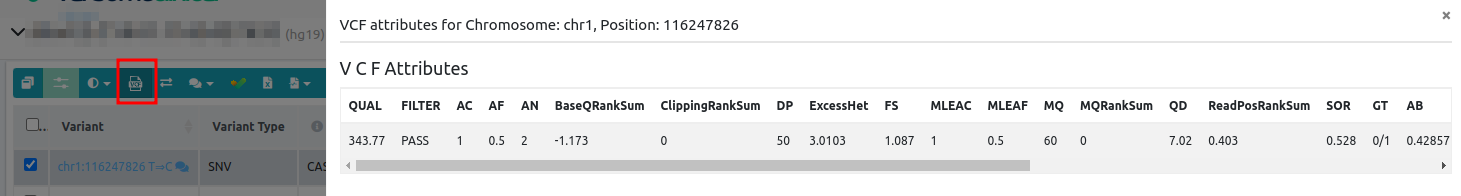
The germline and somatic variant calling pipelines use different algorithms for variant calling and, therefore, the VCFs obtained contain different attributes. In the following lines we will describe the VCF attributes for germline and somatic VCFs.
Germline VCF
- QUAL: it is the phred-scaled probability that the site has no variant. This quality value is used in the pre-processing step (before the annotation) to decide which variants have a PASS call status and which have a FAIL.
- FILTER: filters that have been applied to the variant.
- AC: allele count in genotypes, for each ALT (alternative) allele, in the same order as listed
- AF: allele frequency for each ALT allele in the same order as listed.
- AN: total number of alleles in called genotypes.
- BaseQRankSum: a z-score for base qualities of reference and alternative alleles. For example, a BaseQRankSum close to 0 means the reference and alternative alleles have the same base qualities and a BaseQRankSum around 2 means they differ by 2 SDs. A positive value of 2 means that ALT alleles have higher qualities than REF (reference).
- ClippingRankSum: Z-score From Wilcoxon rank sum test of ALT vs. REF number of hard clipped bases.
- DP: approximate read depth; some reads may have been filtered.
- ExcessHet: phred-scaled p-value for exact test of excess heterozygosity.
- FS: Fisher strand. It is a measure of sequencing bias. This measures if one strand is preferred than the other when sequencing. Larger values means larger bias.
- MLEAC: maximum likelihood expectation of AC (Allele counts).
- MLEAF: maximum likelihood expectation of AF (Allele Frequency).
- MQ: mapping quality. Comparison quality value.
- MQRankSum: this is the u-based z-approximation from the Rank Sum Test for mapping qualities. It compares the mapping qualities of the reads supporting the reference allele and the alternate allele.
- QD: QUAL normalized by read-depth (QUAL/DP).
- ReadPosRankSum: this is the u-based z-approximation from the Rank Sum Test for site position within reads. It compares whether the positions of the REF and ALT alleles are different within the reads.
- SOR (StrandOddsRatio): this is another way to estimate strand bias using a test similar to the symmetric odds ratio test. FS tends to penalize variants that occur at the ends of exons and SOR do not. Reads at the ends of exons tend to only be covered by reads in one direction and FS gives those variants a bad score. SOR will take into account the ratios of reads that cover both alleles.
- GT: genotype. It is encoded as allele values separated by either of / (not phased) or | (phased). The allele values are 0 for the reference allele (what is in the REF field), 1 for the first allele listed in ALT, 2 for the second allele list in ALT and so on.
- AD: allelic depths for the REF and ALT alleles in the order listed.
- GQ: phred-scaled probability that the call is incorrect.
- PGT: physical phasing haplotype information, describing how the alternate alleles are phased in relation to one another.
- PID: physical phasing ID information, where each unique ID within a given sample (but not across samples) connects records within a phasing group.
- PL: normalized, phred-scaled likelihoods for genotypes as defined in the VCF specification.
- SAC: number of reads on the forward and reverse strand supporting each allele (including reference).
- MIN_COVERAGE: minimum coverage threshold considered to give the variant a status of PASS.
- MIN_QUALITY_INDELS: minimum QUAL threshold considered to give the INDEL variant a status of PASS.
- MIN_QUALITY_SNV: minimum QUAL threshold considered to give the variant a status of PASS.
Somatic VCF
- FILTER: filters that have been applied to the variant.
- AS_FilterStatus: filter status for each allele, as assessed by ApplyRecalibration. Note that the VCF filter field will reflect the most lenient/sensitive status across all alleles.
- AS_SB_TABLE: allele-specific forward/reverse read counts for strand bias tests. Includes the reference and alleles separated by a '|'.
- DP: approximate read depth; some reads may have been filtered.
- ECNT: number of events in this haplotype.
- GERMQ: phred-scaled quality that ALT alleles are not germline variants
- MBQ: median base quality.
- MFRL: median fragment length.
- MMQ: median mapping quality.
- MPOS: median distance from end of read.
- POPAF: negative log 10 population allele frequencies of ALT alleles.
- TLOD: log 10 likelihood ratio score of variant existing versus not existing.
- GT: genotype, encoded as allele values separated by either of / (not phased) or | (phased). The allele values are 0 for the reference allele (what is in the REF field), 1 for the first allele listed in ALT, 2 for the second allele list in ALT and so on.
- AD: allelic depths for the REF and ALT alleles in the order listed.
- AF: allele fractions of alternate alleles in the tumor.
- F1R2: count of reads in F1R2 pair orientation supporting each allele.
- F2R1: count of reads in F2R1 pair orientation supporting each allele.
- SB: per-sample component statistics which comprise the Fisher's Exact Test to detect strand bias.
- MIN_COVERAGE: minimum coverage threshold considered to give the variant a status of PASS.
- MIN_QUALITY_INDELS: minimum QUAL threshold considered to give the INDEL variant a status of PASS.
- MIN_QUALITY_SNV: minimum QUAL threshold considered to give the variant a status of PASS.