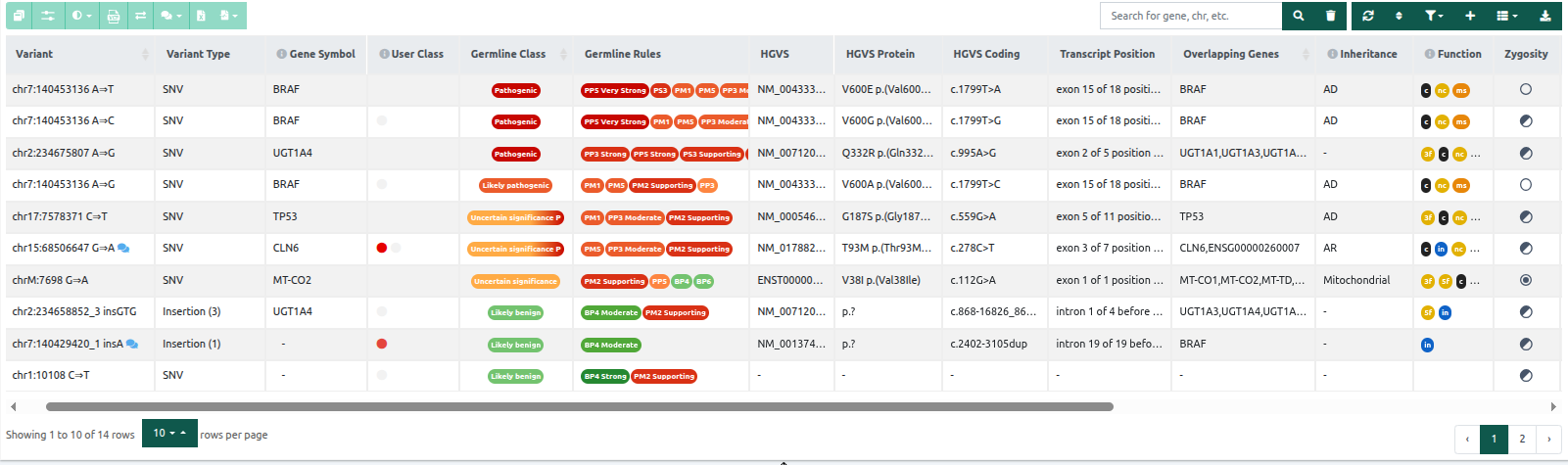
The results are displayed in the Variant Table. Rows contain the identified variants, and columns contain core annotations for each variant (Variant, Variant type, Class, Genes, Function, Zygosity, Allelic balance, and Coverage). However, none of the columns are mandatory - you can choose which ones will be displayed with the "Show/hide columns" icon ![]() . The length of each column on the variant table can be adjusted by dragging the sides of the column headers.
. The length of each column on the variant table can be adjusted by dragging the sides of the column headers.
The column order in the variant table is user-specific, meaning each user can set up a custom order and visibility of columns.
You can hover over the info icon next to the column names to display the column information. The variant table is sorted by the Germline Variant Classification by default showing the most Pathogenic variants at the top. You can use the
![]() icon to sort the variant table by other values (e.g. phenotypes, variant position, coverage) in ascending or descending order. Use the “Reset/refresh” icon
icon to sort the variant table by other values (e.g. phenotypes, variant position, coverage) in ascending or descending order. Use the “Reset/refresh” icon to return the variant table to its original state.
The variant table can be accessed by the user who requested the analysis or by other people belonging to the same group.
Description of results page functionalities
Columns for Germline/Somatic samples
- Variant: The variant’s sequence and genomic location.
- Variant type: SNV (single nucleotide variant); for INDELs and substitutions, the number of nucleotides affected is shown.
- Gene Symbol: Gene used for annotation and classification of the variant for ACMG (& AMP for somatic samples).
- User Class: custom classification for variants. User classifications are also available for AMP and ACMG rules when the user clicks on "Save as manual classification" below the ACMG/AMP verdict. The custom classifications are linked to the variant and will be displayed in other analyses of your group if the same variant is found. See more here: Custom Variant Classifications and Import classifications and comments.
- CNV: this column is shown when the user runs a CNV analysis with the sample. It will have green ticks if any of the following conditions meet:
- there is a CNV that overlaps with the position of the current variant
- there is a CNV in the same gene where the current variant was found (not necessarily in the same position of the variant) - Germline Class: Variants are ordered by our pathogenicity classification:
- 5 = Pathogenic
- 4 = Likely pathogenic
- 3 = Uncertain significance
- 2 = Likely benign
- 1 = Benign.
- Germline Rules: The set of triggered ACMG rules are displayed in clickable bubble icons that include the rule’s description and explanation for triggering.
- HGVS: HGVS nomenclature for the variant.
- HGVS Protein: HGVS nomenclature for the protein sequence change compared to the reference.
- HGVS Coding: HGVS nomenclature for the variant.
- Transcript position: Variant described on the DNA level in relation to a specific gene based on the coding DNA reference sequence.
- Overlapping Genes: The name of any gene(s) the variant falls within.
- Inheritance: Mode of inheritance of the gene from the CGD, OMIM, ClinGen Disease Validity, gene2Phenotype, GenCC and Domino databases:
- AD: autosomal dominant
- AR: autosomal recessive
- XL: X-linked
- BG: blood group
- Function: The position of the variant with respect to the gene it falls within, and its coding effect (if any).

⚠️Please note that in the Function column, we display all possible functional annotations derived from the transcripts listed in the Transcripts tab.
For example, a variant may be an intronic in most transcripts. However, if at least one transcript indicates in the Location column that the variant falls within a 3' UTR, then the variant will be labeled in the Function column as both intronic and 3' UTR.
This approach ensures that potentially relevant transcripts are not overlooked.
VarSome Clinical allows the use of custom transcripts for annotation. For more information please refer to: Custom transcript for annotation.
- Gene Symbol: Gene used for annotation and classification of the variant for ACMG (& AMP for somatic samples).
- Zygosity:

If the variant did not pass the variant caller quality filter, (see FAQs), the zygosity is shown in the table as (failed quality/non- genotyped).
- Phasing group: this column indicates the phasing group of the variant if it exists. Note that this column will be available only for those variants that are phased. If you would like to filter the variant table and visualize only the variants that share the shame phasing group with your variant of interest, you can click on the following icon . If you would like to know more about phasing please go here.
- Allelic balance: Proportion of reads that support the variant. For example, if a variant’s location is covered by 100 reads, of which 25 support the variant and 75 do not, then the variant would have an allelic balance of 25/100 = 0.25.
- Frequency: Frequency of the variant in the general population or (if applied) the specified ethnicity.
- Coverage: Number of reads that align to the variant’s position. For analyses of fastq samples, the blue numbers contain links to JBbrowse, showing the read alignments at the variant’s position.
- Filters: filters that have been applied to the data. Filters are associated with the variant calling quality filters that have been applied to the variant to decide whether it has a call status of PASS or FAIL.
- rsID: the reference SNP cluster ID of this variant.
- Phenotype (if the user selects to add phenotypes): This column will contain the number of user input phenotype(s) associated with the variant gene. More information can be found in the document Phenotype Matching.
Extra columns for Somatic samples
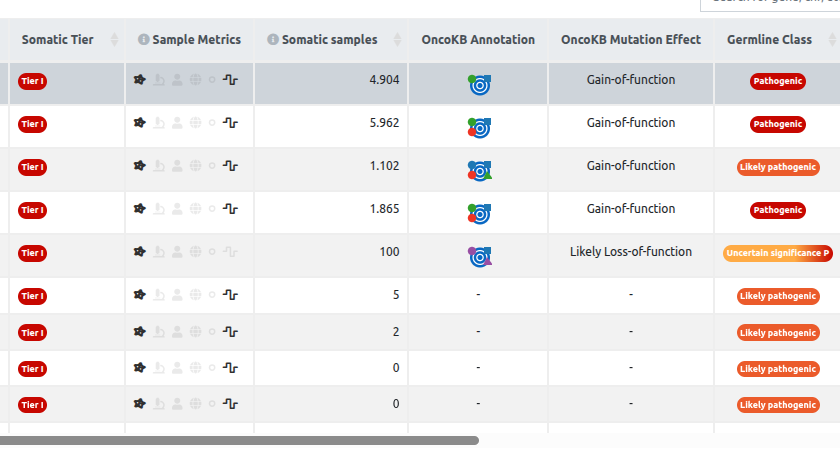
- Somatic Tier: Variants are ordered by an aggregate AMP score (see AMP Implementation documentation for more details), from most pathogenic to benign. Tier I = Cancer with approved drug therapies, Tier II = Cancer but no approved therapy, Tier III = Uncertain Significance, Tier IV = Benign or not related to cancer.
- Sample Metrics: Each icon represents the sample information introduced by the user. They light up when there is data in one of the cancer-related databases matching the variant to the relevant sample characteristic:
- Cancer Match: this highlights any variants for which evidence is found linking to the same cancer type as the sample.
- Tissue Match: This will highlight any evidence associating the variant or gene with the sample tissue.
- Age: This will display the patient's age relative to an age histogram for certain cancer types.
- Ethnicity: It will report the variant's frequency in the relevant ethnic group.
- Sex: more than 50% of reported cases across somatic sample databases match the sample’s sex.
- Variant Allele Frequency: variants with a low VAF are most likely tumor variants, whilst VAFs of 50% and 100% indicate germline variants.
- Somatic Samples: Sum of available affected samples from databases included in Cancer Aggregator (COSMIC, CBioPortal, Cancer HotSpots, GDC).
- OncoKB Annotation: it displays the oncogenicity and the levels of therapeutic and diagnostic evidence of a specific mutation.

- OncoKB Mutation Effect: it displays the functional impact of the mutation at the biological level, independent of its clinical actionability.
For the CNV/SV analysis Variant Table please check the document CNV/SV Variant Table and Cards.
For the Repeat Expansion analysis Variant Table please check the document Repeat Expansion Variant Table and Cards.
For the Fusion analysis table please check the document Fusion Table and Cards.