What is variant calling?
Variant calling is the process by which a software program (the variant caller) identifies variants from sequence data. For germline samples sequenced using hybridization-based capture kits, we use Sentieon's DNA Scope variant caller and for somatic samples, we use the Sentieon TNhaplotyper2 algorithm. On the other hand, for amplicon kits on both germline and somatic samples VarSome Clinical uses VarDict.
Which variant callers are used in VarSome clinical?
Depending on the type of assay used to sequence the sample and on the sample itself, VarSome Clinical uses different variant callers.
VarDict is a sensitive variant caller and is especially well-suited to amplicon samples. VarDict implements several novel features such as strand bias aware variant calling from targeted sequencing experiments.
What is the call status of a variant?
There are different quality metrics associated with each variant which can be used in subsequent steps of the pipeline to assign it a call status. The call status of a variant can be:
- PASS: all the quality metrics are above the thresholds (i.e. the variant has passed all quality filters).
- FAIL: the variant has not passed all quality filters.
- Missing values are specified with a dot (‘.’).
Quality filters
The quality filters used for germline and somatic analyses are different since we use different variant callers, which, in turn, use different parametrization. The parameters used in Sentieon have been optimized for the detection of variants from the GIAB set. In VarDict we currently use a set of minimally changed default parameters, adjusted through exchange with VarSome Clinical users.
Quality filters for capture kit samples
Germline analyses
Sentieon's DNAScope
This caller is used for germline capture kit samples and performs an improved version of GATK Haplotype variant calling. We apply the following quality filters after the variant calling step:
- Coverage: number of reads aligned against the variant position. The minimum coverage for capture kit samples is 8; all variants with coverage lower than 8 reads will be considered as FAIL.
-
Machine Learning (ML) refinement For germline analyses, the pipeline utilizes Sentieon’s DNAscope Machine Learning model. This step applies a pre-trained model to evaluate variants based on complex signatures that standard algorithms may miss (see https://github.com/Sentieon/sentieon-dnascope-ml).
-
MLrejected: This filter tag is applied to variants that have been flagged by the Machine Learning model as low-confidence or probable artifacts.
-
- Single sample analyses: we assign a FAIL call status to the variants having a QUAL lower than 100 in single sample analyses. The QUAL is the Phred-scaled probability that a REF/ALT polymorphism exists at this site given the sequencing data.
-
ReadPosRankSum: compares the position of the alternative allele within the reads carrying the variant versus the position of the reference allele within the reads carrying the reference. Strongly negative values indicate that the alternative allele is found primarily at the ends of reads, which is often indicative of an alignment artifact or False Positive.
-
Threshold: Fails if value is < -8.0.
-
- Multisample analyses (couple, family trio or generic multisample): we use the GQ (genotype quality) which represents the Phred-scaled confidence that the genotype assignment (GT) is correct. All variants with a GQ lower than 20 will be marked as FAIL. Please bear in mind that the GQ is associated with each sample. For example, a variant called in a trio analysis will have three different GQs, one per each sample. The variant might have a GQ below the threshold in one of the samples while having a GQ above of it in the other samples. In that case, the variant will be marked as "Failed/Not genotyped" in the sample where it had a low GQ and PASS in the others.
Somatic analyses
Sentieon’s Tnhaplotyper2
Tnhaplotyper2, which is used for somatic capture kit samples, is designed to behave like GATK’s Mutect2. Tnhaplotyper2, like mutect2, has associated filtering tools which are applied to the variants found by the caller. These filters can then be used to decide whether a variant should be marked as PASS or FAIL. If a variant fails any of the filters present in the “FAIL” column of the table below, it will be marked as FAIL. Failing to pass a filter in the “PASS” column will not cause the variant to be marked as FAIL.
| PASS | FAIL |
|
clustered_events |
map_qual |
|
duplicate |
base_qual |
|
fragment |
contamination |
|
multiallelic |
weak_evidence |
|
n_ratio |
low_allele_frac |
|
orientation |
normal_artifact |
|
position |
panel_of_normals |
|
slippage |
strand_bias |
|
haplotype |
|
|
germline |
|
|
strict_strand |
Somatic VCF filters that do not mark a variant as FAIL:
- clustered_events: multiple events are present on the same haplotype as the variant which is indicative of a false-positive call.
- duplicate: the alternate allele is overrepresented by apparent sequencing duplicates.
- fragment: a large difference is observed in the median fragment length for reads supporting the reference and alternate alleles.
- multiallelic: the mutation occurs at a multiallelic site.
- n_ratio: too many 'N' bases at the site.
- orientation: the variant is likely an artifact due to orientation bias.
- position: the allele is close to the ends of the reads.
- slippage: the variant is likely an artifact due to polymerase slippage.
- haplotype: variant is on the same haplotype as other filtered variants.
- germline: there is evidence that the variant is germline.
- strict_strand: evidence for the alternate allele is not significant on both directions.
Somatic VCF filters that mark a variant as FAIL:
- map_qual: the median mapping quality of reads supporting the alternate allele is too low.
- base_qual: the median base quality of bases supporting the alternate allele is too low
- contamination: the alternate allele is present due to contamination.
- weak_evidence: the mutation does not have significant support above noise.
- low_allele_frac: the variant allele fraction is below the threshold.
- normal_artifact: the variant is likely an artifact in the normal sample.
- panel_of_normals: the site is present in the panel of normals.
- strand_bias: evidence for the alternate allele comes from only one read direction.
Quality filters for amplicon kit samples
Both somatic and germline amplicon kit samples are analyzed using VarDict with different quality thresholds.
Germline analyses
- The Allelic Balance (AB) cutoff is set to 0.2. However this rule applies only for positions covered by more than 100 reads. Otherwise the variant is reported only if the AB is < 20/coverage depth. (I.e. the call will not be made if the variant is supported by less than 20 reads.)
Somatic analyses
- The Allelic Balance cutoff is 0.005. However this rule applies only for positions covered by more than 400 reads. Otherwise the variant is reported only if the AB is < 20/coverage depth. (I.e. the call will not be made if the variant is supported by less than 20 reads.)
Call status variant filtering
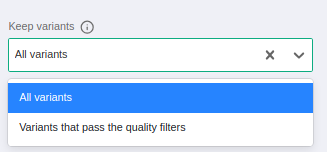
When launching a germline/somatic analysis from FASTQ, the user will have two options:
- All variants: the variant table will contain all variants called by the variant caller including both variants with PASS and FAIL call status.
- Variants that pass the quality filters: the variant table will contain only variants having a call status of PASS.
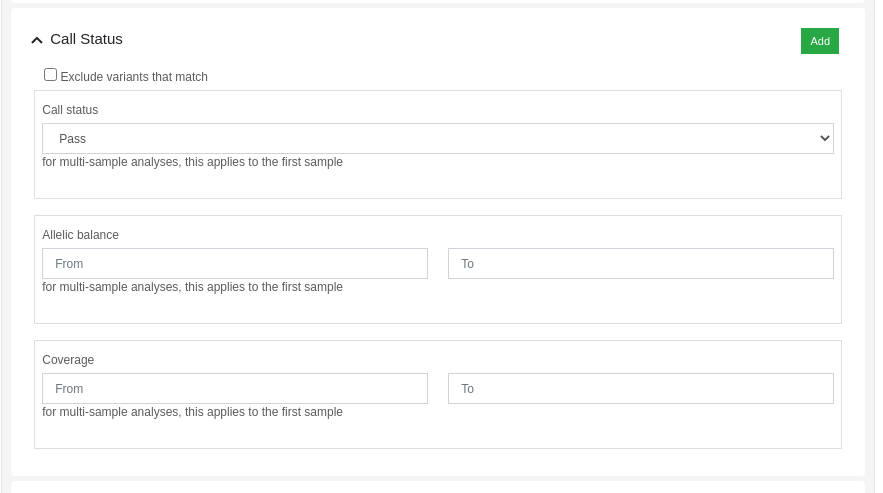
Variants can be filtered by its call status using the dynamic filters feature. The "Call Status" filter allows the user to filter variants based on the following criteria:
- Call Status: PASS, FAIL or anything.
- Allelic balance: proportion of reads supporting the alternative allele.
- Coverage: number of aligned reads against the variant position.