1. ACMG Actionable Genes
Variants are reported in genes listed in the ACMG SF v3.2, classified as pathogenic or likely pathogenic, except for variants in the AR (autosomal recessive) genes.
For AR genes, any pathogenic or likely pathogenic variants are reported only if:
- They are homozygous OR
- There are at least two heterozygous pathogenic or likely pathogenic variants in the gene.
Criteria described in ACMG SF v3.2: Miller et al., Genetics in Medicine, (2023) 25(8), 100866 (DOI: 10.1016/j.gim.2023.100866).
2. Carrier risk for couple
Pathogenic variants for which both members of a healthy couple are heterozygous and so can be passed on to any offspring. Included are genomic (not mitochondrial) variants that are classed as pathogenic, likely pathogenic, or of uncertain significance, are not homozygous in either prospective parent and either:
- Both individuals are heterozygous for the variant
- The variant is on the X chromosome of the mother
- The variant falls in a gene that has at least two variants where one is present and heterozygous only in one individual and the other is present and heterozygous only in the other individual. In such cases, all pathogenic, heterozygous variants for that gene are shown as they are candidates for compound heterozygosity.
Options:
- Strong VUS: If selected, keep variants of Unknown Significance (but only if one of the strong pathogenic Germline rules has fired for this variant). VUS variants qualify only if they trigger one of the Germline rules: PVS*, PS*, or PP5. If not selected, keep all VUS variants (irrespective of Germline rules).
Strong rules are PVS1, PS1, PS2, PS3, PS4 and PP5. In addition, any rule whose strength has been raised to "strong", "very strong" or "stand alone" will be considered a "strong" rule, even if its original strength as per the Germline guidelines was lower. For example, PM1 can be raised from "supporting" to "strong" if the variant is located in a dense hot spot.
We recommend you further filter by genes with a recessive mode of inheritance or loss-of-function pathogenicity.
3. Compound Heterozygous Candidates
Variants classified as pathogenic, likely pathogenic, or of unknown significance for which all of the following apply:
- they are heterozygous variants in genes that carry at least one other heterozygous variant and no homozygous pathogenic variants.
- are not in mitochondria
Options:
- Strong VUS: If selected, keep variants of Unknown Significance (but only if one of the strong pathogenic Germline rules has fired for this variant). VUS variants qualify only if they trigger one of the Germline rules: PVS*, PS* or PP5. If not selected, keep all VUS variants (irrespective of Germline rules).
- Homozygous also: If selected it will also filter for homozygous variants.
- Phased Mode: If selected, it will only filter those variants with phasing information to identify compound heterozygous variant pairs in the same gene and phasing group and in different zygosities (1|0 vs 0|1).
4. Compound Heterozygous for Trios (n=3)
This filter is aimed to support the identification of compound heterozygous variants in an affected child when the genome of the two unaffected parents is also provided (Family Trio analysis (n=3)).
The filter will keep variants in the child classified as pathogenic, likely pathogenic, or of unknown significance for which all of the following apply:
- They are heterozygous variants in genes that carry at least one other heterozygous variant and no homozygous pathogenic variants.
- Are not in mitochondria
If such variant pairs are detected, it will look for compound heterozygous pairs in the same gene in each of the parents.
If either parent does have a compound heterozygous pair in the same gene (not necessarily the same pair as the child), then we discard the pair identified in Step A and move to the next candidate. The assumption is that the parents are unaffected, so the child would also be unaffected if one of the parents has a compound heterozygous pair in the same gene.
Options:
- Use phasing information (phased mode). When this option is enabled the filter will identify compound heterozygous variant pairs in the child—that is heterozygous variants in the same gene and phasing group and in different zygosities (1|0 vs 0|1).
- Strong VUS: If selected, keep variants of Unknown Significance (but only if one of the strong pathogenic ACMG rules has fired for this variant). VUS variants qualify only if they trigger one of the ACMG rules: PVS*, PS*, or PP5. If not selected, keep all VUS variants (irrespective of ACMG rules). Strong rules are PVS1, PS1, PS2, PS3, PS4 and PP5. In addition, any rule whose strength has been raised to "strong", "very strong" or "stand alone" will be considered a "strong" rule, even if its original strength as per the ACMG guidelines was lower. For example, PM1 can be raised from "supporting" to "strong" if the variant is located in a dense hot spot.
- Homozygous also: If selected it will also filter for homozygous variants.
5. De novo (strict)
Variants present in the proband and absent in both parents, where neither parent has any reads supporting the variant but only counting positions where the parents have a minimum coverage of 8.
6. De novo candidates (naive)
Variants likely to have arisen in the child from unaffected parents. Variants must meet either of the following conditions:
- the child is homozygous for the variant, but the variant is only present in one parent OR
- the variant is present in the child but not present in either parent.
We recommend you to further filter for pathogenicity, coverage, frequency, and mode of inheritance.
7. Exonic & Splicing algorithmic filter
A special case of algorithmic filter is “Exonic and splicing” which only keeps exonic (including UTR and other non-coding exons) and splicing (no more than 10 nucleotides from a known splice site) variants.
The filter may be launched manually, just like any other, but it will also be run automatically on any analyses with more than 500,000 variants.
This algorithmic filter provides the same results as would occur if you made a dynamic filter with the following criteria:
- Coding
- Splicing
- Non-coding exon +3’ UTR
- Non-coding exon +5’ UTR

The aim of this filter, and the reason it will run automatically for large analyses, is to provide a smaller subset of results to the user which will be far quicker and easier to sort through. Since, even with WGS analyses, the variants of interest tend to be those that can affect the protein sequence, we feel that this filter will help our users quickly identify and focus on the variants of interest even on larger samples such as WGS.
⚠️ Please note that while the filter will be run automatically for such large analyses, the full result set will still be available as usual. The filter will run as a sub-analysis and will not affect the results of the main, parent analysis in any way.
Later on, you lay over Dynamic filters to fine-tune the list of your variants.
8. Family trio recessive (coding and rare)
The following criteria are applied in this order.
- Keep only either:
a. Coding variants that are frameshift or missense or nonsense or stop-loss or exon deletion or in frame or start loss or splice junction loss
b. Splicing variants (+/-10 bp from exon ends)
- Remove variants where allelic balance < 0.2 or coverage <= 5.
- Remove variants present in both parents except if the child is homozygous and both parents are heterozygous.
- Remove variants with gnomAD population frequency over 1% based on the ethnicity of the proband. If ethnicity is not provided, the general population frequency is used.
- Remove variants that are homozygous for the alternative allele in either parent.
- Remove variants in mitochondria and chromosome Y.
- Keep only variants where EITHER of these criteria applies:
b. Child is heterozygous, and the following criteria BOTH apply:
-
- There are two or more variants in the same gene. To qualify, a variant must be in a coding transcript of a gene with a Transcript Support Level consistent with the sample’s settings. The variant must be Pathogenic, Likely Pathogenic, or of Uncertain Significance (VUS).
- The variants did not all come from the same parent; some variants on the gene may have come from the mother and some from the father, or are de novo.
Filter assumptions:
- This multi-sample consists of children and parents.
- The last sample is the father, and the one-from-last sample is the mother. The last two samples must be unaffected; otherwise, the filter returns an error.
- All affected children are tagged as affected, and all unaffected are tagged as unaffected; there is at least one affected child; otherwise, the filter returns an error.
This filter is not applicable to and should not be run for multi-samples that don’t comply with the above.
Options: All of the following fixed values originally set for the filter can now be changed:
- Maximum distance from the splice site
- Minimum distance from the splice site
- Minimum coverage
- Minimum allelic balance
- Maximum frequency
- Include all variants (i.e., B, LB, VUS, LP, and P). If not selected, only variants classified as VUS, LP, or P will be included.
This filter specification was kindly contributed by Dr Erica Davis of Lurie Children’s Hospital in 2021.
Please note that this algorithmic filter can be used for three or more samples.
9. Fisher exact test
This filter will select any variants that are found more often (fisher exact test p-value <=0.05) in the affected samples than in the controls.
10. Genes in common
All variants classified as pathogenic, likely pathogenic, or of unknown significance (but only if one of the strong pathogenic Germline rules has fired for this variant), which are found in genes with at least one such variant in all samples of a merged analysis.
The filter works in the following way:
- It will identify all genes with at least one pathogenic, likely pathogenic, or VUS variant in all samples.
- It will then return ALL pathogenic, likely pathogenic, or VUS variants falling in that set of genes.
- Pathogenic only: If this option is selected, variants classified as Benign(B), Likely Benign(LB), or VUS/LB-B will be excluded from the results.
11. GWAS Catalog
All variants with GWAS Catalog data.
Options:
- Pathogenic only: If this option is selected, variants classified as Benign(B), Likely Benign(LB), or VUS/LB-B will be excluded from the results.
12. Imprinted Genes
Variants classified as pathogenic, likely pathogenic, or of unknown significance which are heterozygous in the proband and either:
- heterozygous in the father and found in a maternally imprinted gene or
- heterozygous in the mother and found in a paternally imprinted gene
Options:
- Pathogenic only: If this option is selected, variants classified as Benign(B), Likely Benign(LB) or VUS/LB-B will be excluded from the results.
13. Max other samples
Remove variants that exist in more than the selected number of other samples from your group, and those with more than 5% population frequency.
14. PharmGKB
Variants associated with genes that have PharmaGKB information. For more information see the section Pharmacogenomics Knowledge Base (PharmGKB).
Options:
- Pathogenic only: If this option is selected, variants classified as Benign(B), Likely Benign(LB), or VUS/LB-B will be excluded from the results.
15. Segregating variants
For some of these filters, in order to add the parameterization functionality we have integrated similarly functioning filters into one. For example, you can now find under the filter “Segregating Variants”, the filters previously known as:
- Segregating Variants (dominant, all VUS),
- Segregating Variants (dominant, strong VUS)
- Segregating Variants (recessive, all VUS)
- Segregating Variants (recessive, strong VUS)
- Compound Heterozygous Segregating Variants (all VUS)
- Compound Heterozygous Segregating Variants (strong VUS).
For more information please see here: Segregating variants analysis.
16. Trio Recessive
Pathogenic variants that may be causative of recessive disorders in the child of unaffected parents.
We follow this selection process:
- We exclude variants that are homozygous for the alternative allele in either parent.
- We exclude variants in mitochondria and chromosome Y.
- We keep variants where EITHER of these criteria apply:
- Variant is homozygous for the alternative allele in the child.
- Child is heterozygous and the following two criteria BOTH apply:
- The variants did not all come from the same parent; some variants on the gene may have come from the mother and some from the father or are de novo.
- There are two or more variants in the same gene. To qualify, a variant must be in a coding transcript of a gene with a Transcript Support Level consistent with the sample’s settings. The variant must be Pathogenic, Likely Pathogenic, or of Uncertain Significance (VUS).
Options:
- Strong VUS: If selected, keep variants of Unknown Significance (but only if one of the strong pathogenic Germline rules has fired for this variant). VUS variants qualify only if they trigger one of the Germline rules: PVS*, PS*, or PP5. If not selected, keep all VUS variants (irrespective of Germline rules).
- Missing from one parent: Only keep variants that are missing from at least one parent (strict); in other words, neither parent has all variants that the child has in the same gene.
- Maximum frequency for recessive: We exclude any variants that are found with a frequency greater than the selected threshold. The default value is set to 1 so that no variants will be excluded based on the frequency.
We want all VUS variants to be reported regardless of the triggered rules and their strength, so the "Strong VUS" option will not be selected. We also have the choice to keep only the variants that are missing from at least one parent by selecting the "Missing from one parent" option and we can exclude any variants that are found with a frequency greater than the selected threshold.
![]()

Once the parameters of the filter are set we click on the "Save" button and then on the  on the bottom of the page.
on the bottom of the page.
We recommend you to further filter by genes with recessive mode of inheritance or loss-of-function pathogenicity.
From 19 variants of the initial data set, we end up with 10 variants passing the filter.
Initial analysis variant table:
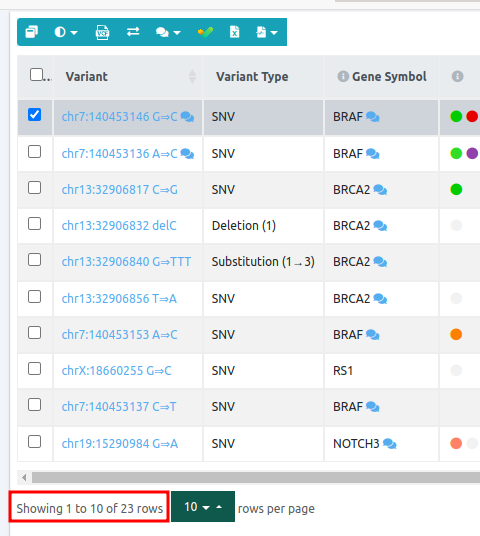
Sub analysis using the "Trio Recessive - All VUS" filter variant table: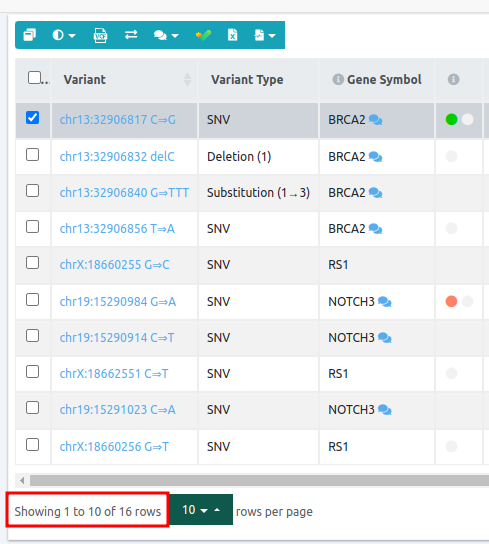
We applied a filter for Autosomal mode of inheritance (Dominant and Recessive) and we ended up with 10 candidate variants.


17. Variants in Common
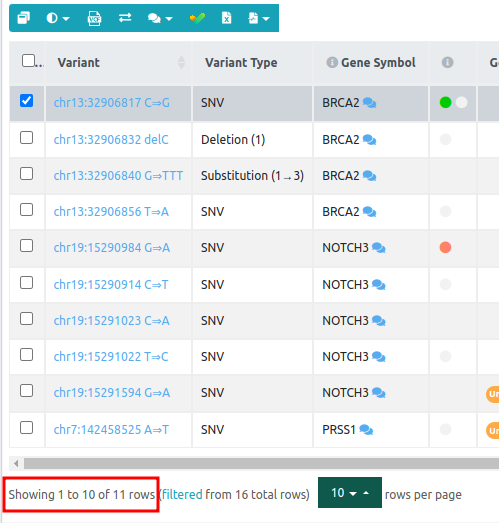
Variants annotated as pathogenic, likely pathogenic, or of unknown function (but only if one of the strong pathogenic ACMG rules has fired for this variant) that are present (in any zygosity) in all samples.
Options:
- Common only to all affected: If selected, keep all variants that are found (in any zygosity) in all affected samples; their status in non-affected samples is ignored.
- Pathogenic only: If this option is selected, variants classified as Benign or Likely Benign will be excluded from the results.
18. VarSome Picks
VarSome Picks is an advanced algorithmic filter, designed to empower bioinformatic analysis of genomic variants using AI. This tool goes beyond conventional variant prioritization methods by considering essential parameters such as phenotype, gene, and variant data, all within a disease-specific context. Its primary objective is to rank potentially causative variants, aiding researchers and clinicians in identifying significant genetic associations related to specific diseases.
You can find more information about VarSome Picks here.
Algorithmic Filters offer a great deal of flexibility and can be fully adjusted according to your needs and your specific workflow. We can encode practically any kind of filtering criteria.
⚠️ Please note that depending on the complexity of the desired filter the setup process may be subject to a fee.