When the sample is associated with phenotypes, variants found in genes linked to at least one of the phenotypes (referred to as top candidate genes) will be prioritized.
Additionally, the top candidate genes should adhere to the mode of inheritance characteristic of the provided phenotype. For instance, if a gene exhibits different modes of inheritance for different phenotypes, only the mode of inheritance linked to the phenotype(s) associated with the sample will be considered. Therefore, the filter will retain the variants that meet any of these criteria.
1. Variants found in top candidate genes that have at least one phenotype match with the phenotypes provided by the user.
2. Variants with Germline classification (ACMG) as pathogenic, likely pathogenic, or strong VUS:
- In a heterozygous state, within genes exhibiting an autosomal dominant mode of inheritance.
- Candidates for compound heterozygosity within genes exhibiting an autosomal recessive mode of inheritance.
- In a homozygous state, within genes exhibiting an autosomal recessive mode of inheritance.
- In a heterozygous state, within genes exhibiting an X-linked dominant mode of inheritance.
- In a homozygous state for males, within genes exhibiting an X-linked recessive mode of inheritance.
- Associated with mitochondrial diseases.
- Found in genes from the ACMG gene list (v3.2) for incidental findings.
Please note that variants marked as artifacts (either manually or automatically) in the main analysis will be excluded from the VarSome Picks results.
Main Features
Comprehensive Data Sources
- HPO (Human Phenotype Ontology) - is used to get the phenotype (HPO term) - gene association
- GenCC (Genetic Clinical Characterization)
- CGD (Clinical Genomic Database)
- ClinGen Disease Validity
- MONDO (Monarch Disease Ontology) - is used here to get the disease (MONDO term) - gene associations.
- OMIM (Online Mendelian Inheritance in Man)
- Gene2phenotype
- PanelApp
Please note that there is no single source from which all relevant genes can reliably be mined. As a result, extracting data for gene-disease associations from multiple databases is likely to produce a more comprehensive list of variants found to be causative for the disease. Some of those are likely to have been classified as VUS.
VarSome Picks incorporates crucial genetic and phenotypic parameters to prioritize variants accurately. These include:
- Phenotype(s) Selected by the User: VarSome Picks ranks variants found in the Top 10 genes associated with the selected phenotype(s) by the user. This feature facilitates tailored analysis, focusing on variants with higher disease relevance.
- Zygosity: Variant-related information on zygosity is considered to assess the impact of heterozygous or homozygous variants on the disease phenotype.
- VarSome germline classification: The algorithm applies the ACMG (American College of Medical Genetics and Genomics) criteria for germline variant classification, ensuring robust variant categorization.
- Mode of inheritance: Gene-related information on the mode of inheritance is taken into account to identify variants associated with specific inheritance patterns.
As well as, quality parameters of:
- Allelic Balance: For germline variants, the algorithm applies a threshold for allelic balance to ensure reliable variant calling.
- Coverage in the sample: Coverage information is evaluated to gauge the depth of sequencing and assess data quality.
How to use VarSome Picks
VarSome Picks supports germline analyses from FASTQ files and VCF files against hg38 and hg19 reference genomes. It is also available for gene list analyses.
The filter can be run on-demand (Analysis actions > New algorithm filter analysis) once the main analysis has finished. It can also run automatically for all analyses if the group supervisor enables this option in the analysis preferences.
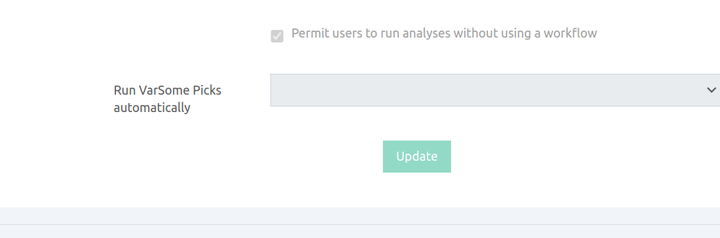
VarSome Picks now runs automatically whenever phenotypes are provided. No manual action is required from the user. This automatic execution applies at two points:
-
If phenotypes are entered during the sample definition step.
-
If phenotypes are added or modified after the analysis has completed.
Whenever phenotypes are added or modified by the user in an existing analysis, if the supervisor has enabled the automatic analyses option, VarSome Picks will re-run automatically. The previous output is erased, ensuring up-to-date and accurate results for the latest phenotype selections.
The results will be shown as the result of any other algorithmic filter, and in this case, with the variants being ordered by the VarSome Pick’s assigned priority.
VarSome Picks runs only for:
- Single sample germline analysis
- Gene lists analysis
- Trio analyses
⚠️Note: The automated execution, is currently available only for Single sample germline analysis and trio analyses. Users should provide the suspected disease and phenotype.
Users should provide the suspected disease and phenotype.
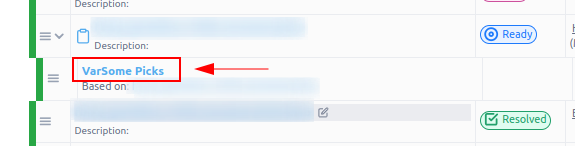
It is also possible to run VarSome Picks even if phenotypes are not initially provided. In this scenario, the algorithmic filter will not be triggered automatically. However, a warning will be displayed to make sure our users are aware of the potential limitations of the analysis due to the absence of phenotype information:

Note to the User/Disclaimer:
Parameters that might affect the accuracy of VarSome Picks:
- Non-specific or highly genetically heterogeneous phenotype/phenotypic expansion.
- Idiosyncrasies in search functionality (e.g. "hearing loss” instead of a query using the term “deafness” may not return the relevant gene).
- Variable expressivity/age-related penetrance which are parameters that the algorithm does NOT take into account.
Therefore, the decision regarding the appropriateness of this approach has to be at the discretion of the experts - the users.