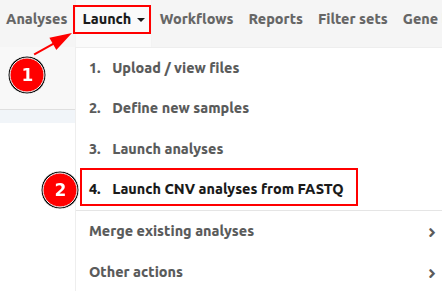
To start a CNV analysis from FASTQ, please go to "Launch" > "4. Launch CNV analyses from FASTQ" as shown in the picture below.
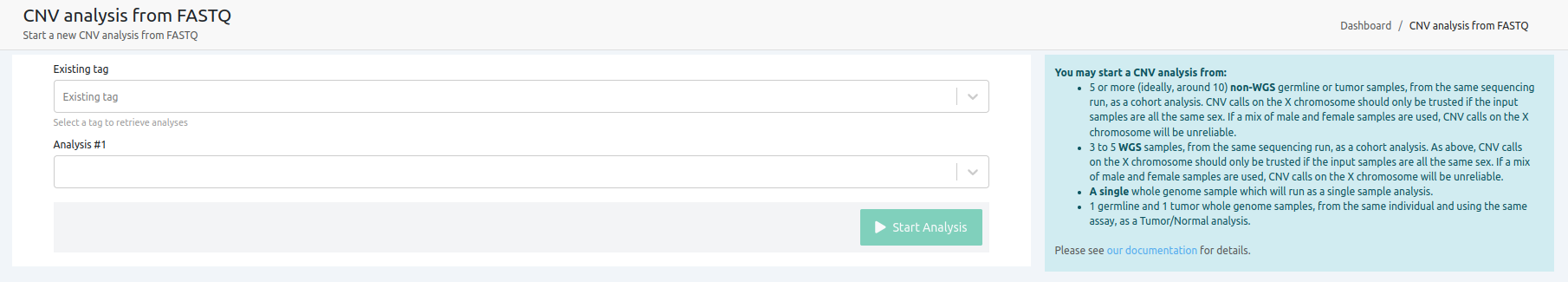
VarSome Clinical interface allows you to select a minimum of 5 and a maximum of 25 already analyzed samples to be used as a cohort for CNV calling. For best results, we recommend you select 5-10 samples from unrelated individuals of the same sex that were sequenced on the same sequencing run.
An alternative method to simplify the process of selecting analyses for CNV calling is to utilize the tagging feature to group your samples. Once grouped, you can directly select the corresponding tag, which can save time and improve accuracy.
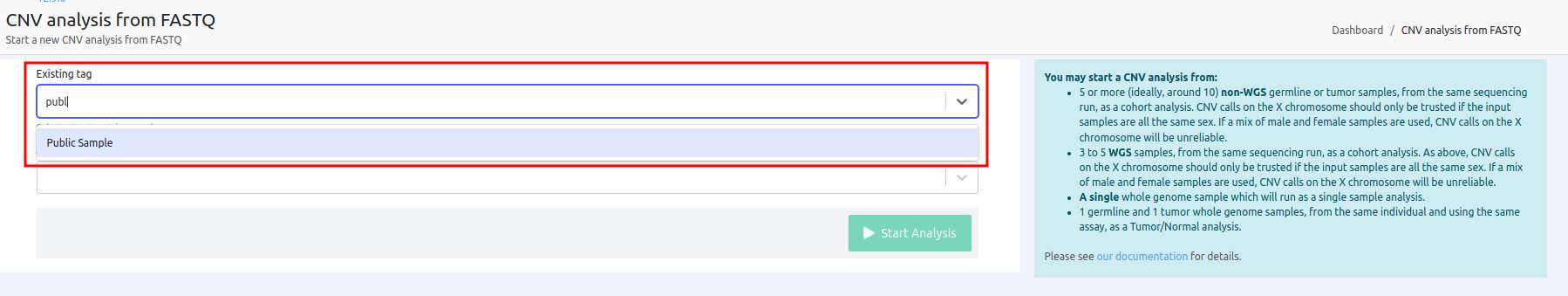
For detailed instructions on how to create and use tags, please refer to the document Sample tags.
Each sample’s results will appear as a sub-analysis of the main analysis.

⚠️ Please note that you can only run CNV tumor-normal analyses if the samples are WGS and samples which have been analyzed using one of the "Generic" kits cannot be used for CNV sub-analyses.
VarSome Clinical currently offers two types of CNV calling solutions from FASTQ:
- ExomeDepth - suitable for cohorts of WES/panels and also for WGS samples.
- Delly - suitable for single WGS samples and WGS tumor-normal CNV analysis.
Whole exome sequencing (WES) or targeted panel data
For the non-WGS analyses we use ExomeDepth, a CNV caller based on a read depth approach. To accurately detect CNVs, ExomeDepth requires at least five samples (ideally around ten) germline or somatic samples that have already been analyzed on VarSome Clinical. They will be run as a cohort with each sample analyzed, using the rest as a pool to select reference samples. The samples should all have been sequenced using the same assay since CNV calls will only be made within the assay's target regions.
For optimal results, the reference set of samples must have the following characteristics in common with the test sample (sample of interest):
- Samples should be prepared with the same library protocol and sequenced by the same sequencing platform.
- All samples (the test sample and the reference) should have been generated as part of the same sequencing batch. It is possible to use samples generated in different batches, but the resulting CNV calls are likely to be much less accurate.
- Samples should originate from individuals unrelated to each other. For example, if samples come from the same family, related individuals should be excluded.
- For CNV calls in sex chromosomes, all samples should be of the same sex (either all male or all female). If they are not all of the same sex, calls on those chromosomes will not be reliable.
All samples are analyzed together and the results (along with a visual display) of each sample are shown as a sub-analysis of that sample.
⚠️ Please note that an inherent limitation of WES is that it produces reads only covering the ~2% of the human genome that falls in exons. Therefore, the full spectrum of CNVs and breakpoints may not be completely characterized. In addition, many large CNVs and cross-chromosome events may not be detected. For optimal results, we suggest either sequencing the entire genome (WGS), or a different experimental approach such as array CGH. Nevertheless, CNV detection based on WES data may give an insight into CNV patterns for a specific disease or phenotype. For more details on the limitations of calling CNVs in such data, please see [1].
Why are reference samples necessary in CNV calling for non-WGS samples?
The read depth approaches used for CNV calling in WGS usually assume that reads distribute in a more or less uniform way across the genome and, therefore, the differences in read depth are used to identify CNVs. However, this assumption fails in the context of WES and targeted sequencing. One of the main reasons is that the probes used for capturing the different targeted regions have variable specificity and efficiency depending on the region. This fact introduces strong biases in the number of mapped reads per region that hamper the CNV detection. ExomeDepth requires multiple samples because it uses them to control the biases given by the extensive variability in capture efficiency across exons and/or target areas.
How are reference samples used by ExomeDepth?
Each sample given as input for ExomeDepth analysis will be taken to call CNVs on it by using a selection of the remaining as reference samples. This means that, when running a CNV analysis with ExomeDepth, you will get calls for all of the input samples, no matter if you consider the sample as test or reference.
Another important key to bear in mind is that every input sample might not be compared to all other samples. Each input sample is compared against an optimized set of reference samples that are well correlated with it. The first step of the CNV calling process is to construct the reference set of samples. To do this, ExomeDepth takes one of the input samples and ranks the remaining by order of coverage correlation with the first sample. Then, the remaining samples are sequentially added to the reference set. After the addition of one sample to the reference set, a statistical calculation is performed to see how good is the current reference set to predict CNVs on the test sample. The addition of samples to the reference set stops when it is unable to improve the reference set power to predict CNVs. Therefore, using a high number of samples for CNV analysis does not necessarily increase the accuracy of the results because:
- Not all available samples are included in the reference set, which means that not all the samples are used as reference for calling CNVs in the test sample.
- Some of the CNVs present in the test sample can be missed if they are shared between the test and the reference samples.
The reference selection process is automated in the analytical pipeline implemented in VarSome Clinical and does not require any additional steps by the user. VarSome Clinical SV pipeline will analyse all samples of the cohort successively and generate CNV calls for all.
ExomeDepth authors estimate that the optimum size of the reference set is ~10. Adding further samples in the reference set actually might decrease the power.
Sensitive mode
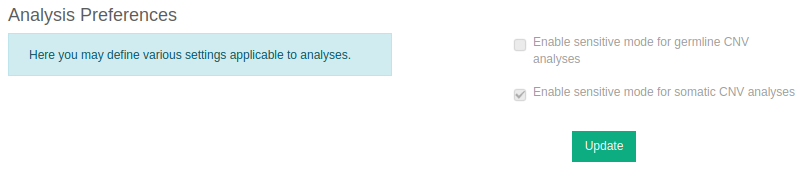
CNV calling for non-WGS CNV analyses is also available in “Sensitive mode”. Compared to standard mode, a lower CNV detection threshold is applied, resulting in more sensitive calling and typically in a higher number of calls. CNV detection can be particularly challenging; for instance single exon CNVs can be hard to call. Still, in a clinical setting, the ability to detect such CNVs is of paramount importance. Sensitive mode is optimised for the needs of clinical laboratories. It allows a shift to the trade-off between recall and false discovery to benefit sensitivity, compared to the standard mode.
You can enable this feature for either somatic or germline samples (or both) in Preferences.
⚠️ Please note that these settings are only available to the group administrator and any changes will be applied to all users of the group.
Whole genome sequencing (WGS) data
For WGS samples, VarSome Clinical offers two solutions:
- CNV calling for a single WGS sample. We use delly, an integrated structural variant (SV) caller tool that can detect both CNVs and other forms of Structural Variants (SVs) at single-nucleotide resolution in short-read genomic sequencing data. It combines 3 different approaches (paired-ends, split-reads and read-depth) to discover extensive genomic rearrangements. Quality passed CNV calls (deletions and duplications) are retained, while other types of SVs are currently not reported.
- CNV calling for a cohort of (2-5) WGS samples. The ExomeDepth caller has been adapted to also process WGS. The solution is suitable for samples with long CNVs (>50kb) that may not be reliably called by Delly. For WGS, the assay target regions comprise the complete genome, split into 50Kb bins. As a result, this imposes a hard minimum size limit: no CNVs smaller than 50Kb can be detected using this approach. The requirements for non-relatedness between the samples and their processing by the same laboratory, sequencer and ideally in the same batch, apply to WGS samples too. All samples are analysed in a single CNV analysis and the results (along with a visual display) of each sample are shown as a sub-analysis.
CNV annotation (from VCF)
Please check the document SV annotation from VCF.
See also
References