VarSome Clinical provides a pipeline to annotate SVs from VCF files. There are two ways to annotate SVs from VCFs.
- Provide a valid VCF file that contains both structural variants (SVs) and SNPs/small INDELs (mixed file) when launching a new analysis either from FASTQ or VCF. Files that include both small variants and SVs will be split to two separate files, one for small variants and one for SVs (i.e .filtered and .cnv).
- Provide a valid VCF file containing only SNPs/small INDELs (small variants) and another VCF that contains SVs (CNVs and/or any other type of SV). The VCF file shall be split into different files:
- VCF containing only small variants
- VCF containing SVs (i.e named filename.sv.vcf.gz)
The SV analysis from VCF is launched as a sub-analysis of the main analysis. You can launch a SV annotation by:
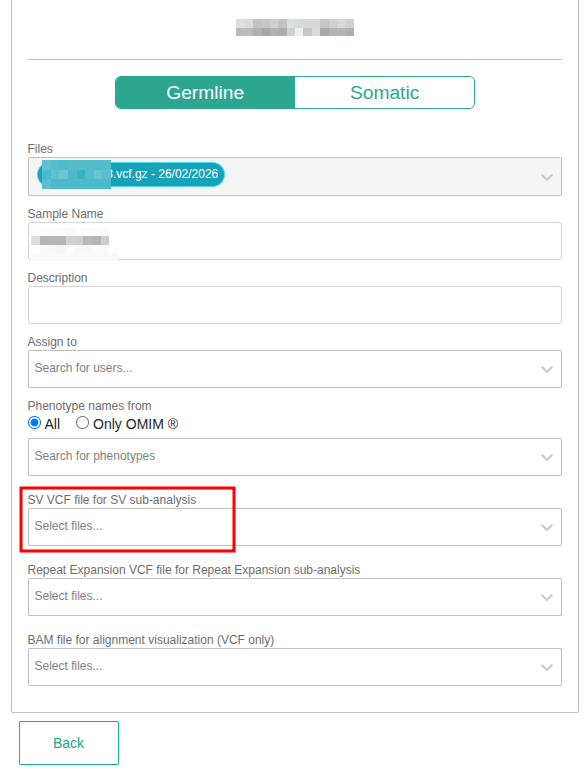
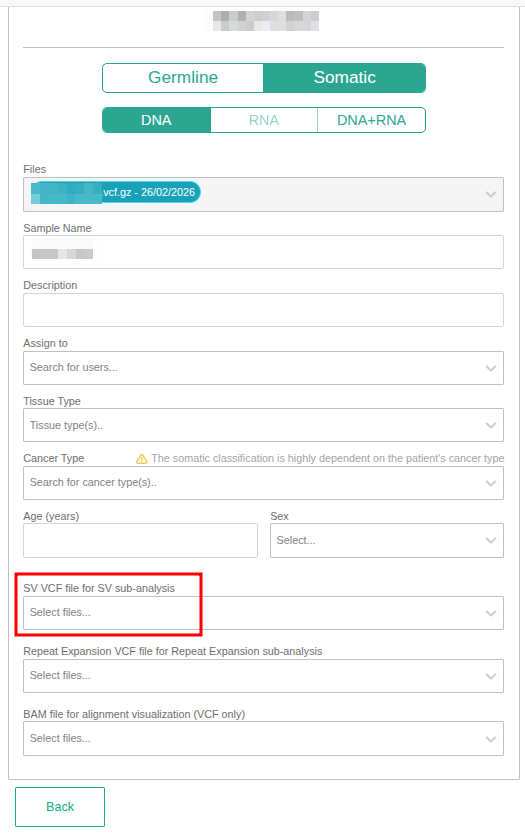
- Adding an SV VCF file when defining your sample.
Germline sample
Somatic sample
- Launching the analysis once the main analysis has finished as a "New SV sub-analysis" either from single or multi sample analyses.
From the Analyses menu:

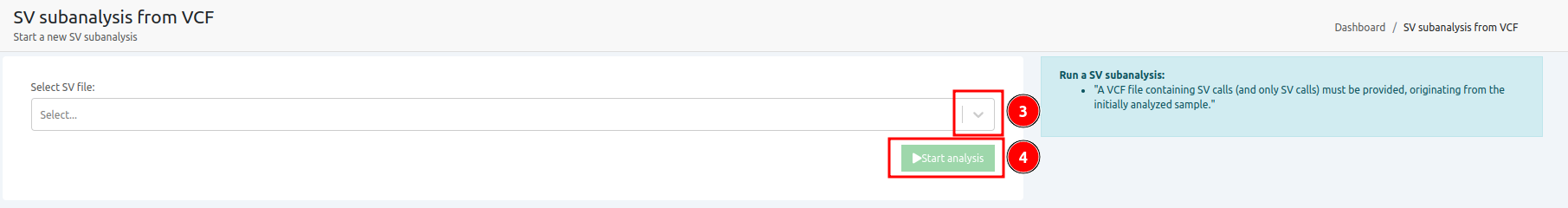
From the Analysis actions menu: 