VarSome Clinical users can attach phenotypes to germline samples. When provided, a new column in the variant table (Phenotypes) is displayed showing the number of associations between the genes and the phenotypes provided. Consistent with the VarSome platform, we have also merged diseases into phenotypes so the users can now attach phenotypes (using HPO terms like before) and diseases (using MONDO and OMIM® terms).
How can I add phenotype(s) to my sample?
Launch analysis page
In the "Launch" > "3. Launch analyses" page you can select "All" or "OMIM" in the respective field
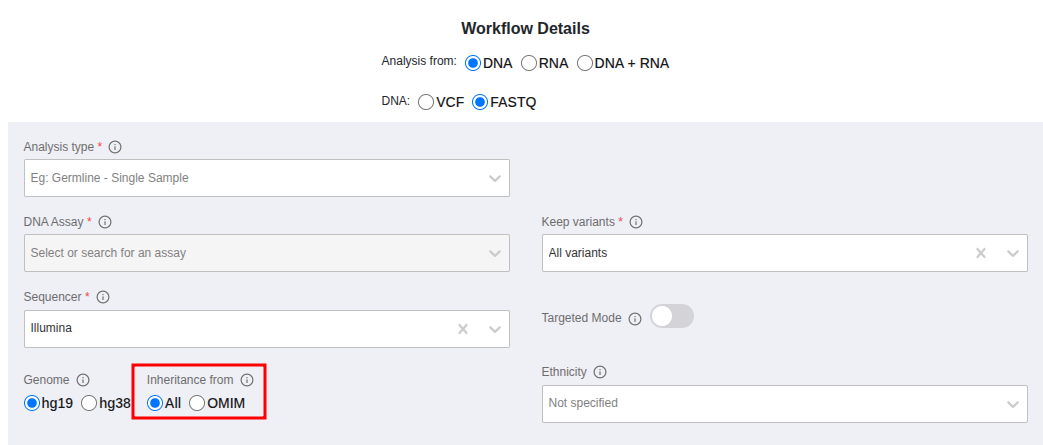
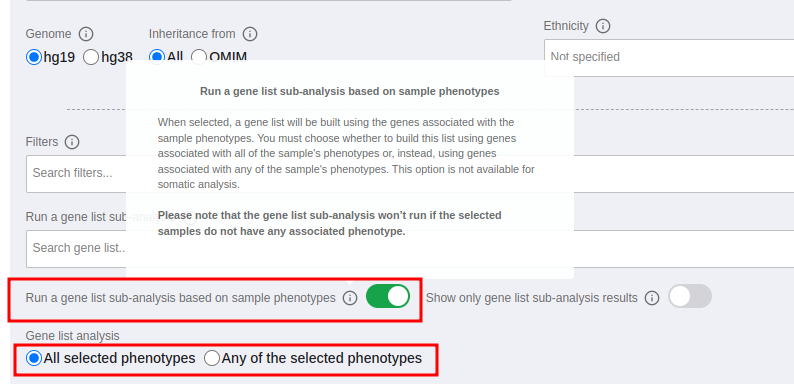
and then you can select to run a gene list sub-analysis based on sample phenotypes. You can either select all the selected phenotypes or any of them as shown in the picture below.
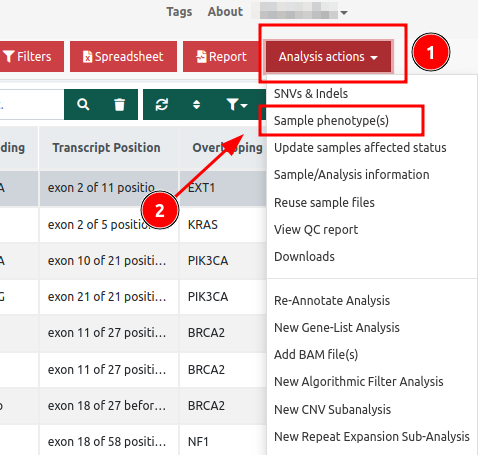
Analyses page - Analysis actions
You can also add phenotypes in an already analyzed sample by clicking on the button of the Analysis actions menu either when viewing your sample in the Analyses page:
Or you can edit the Phenotype information from the results page of an analysis by clicking on the "Analysis actions" button:
You will then be directed to a new screen where you can add the phenotype information. You can begin typing to search for individual terms, or you can bulk input or paste an entire list of HPO, MONDO and OMIM® identifiers (separated by commas, spaces or line breaks), or full phenotype terms (separated by line breaks), directly into the field. The system automatically parses the content and adds all valid entries instantly. Once you are done, click on "Save" to apply the changes. After a few minutes, the "Phenotypes" column will be updated
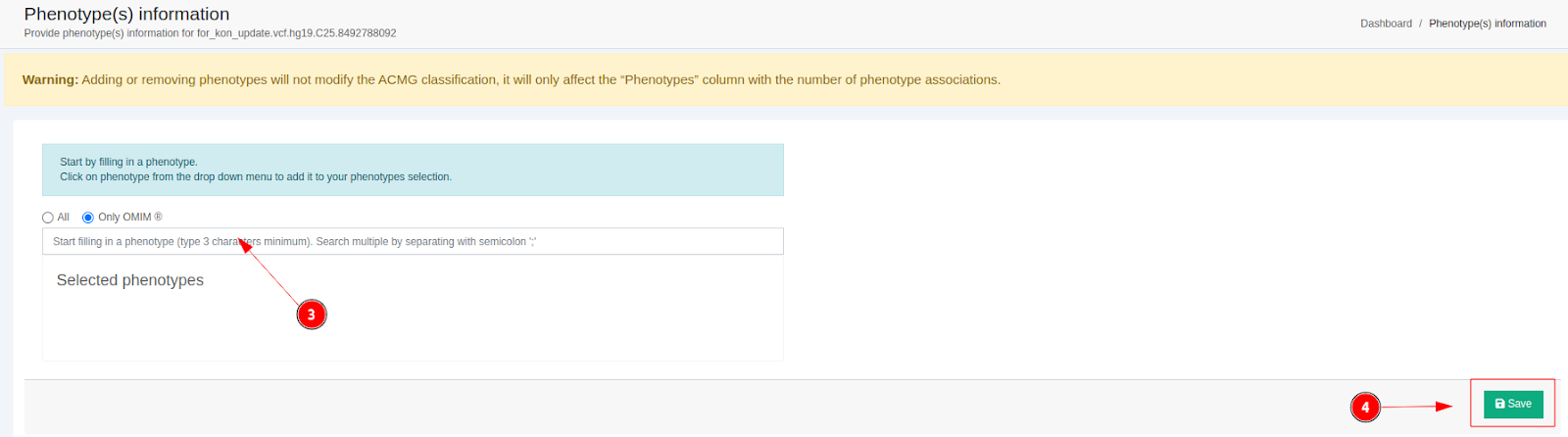
⚠️ Please note you will receive immediate visual feedback indicating which terms were successfully added (blue) and which were unrecognized (red), all while preserving any previously added phenotypes.
There will be a column named "Phenotypes" in the variant table with a value per each variant. This column will contain the number of user input phenotype(s) associated with the variant gene.
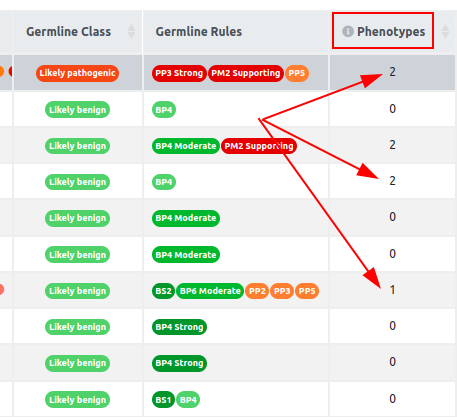
Hover over the number with the mouse to see the name of the matched phenotypes. This column can be used to sort the table in descending/ascending order.
The "Phenotypes" column is also available in CNV analyses. More information can be found in the document SV Variant Table and Cards.
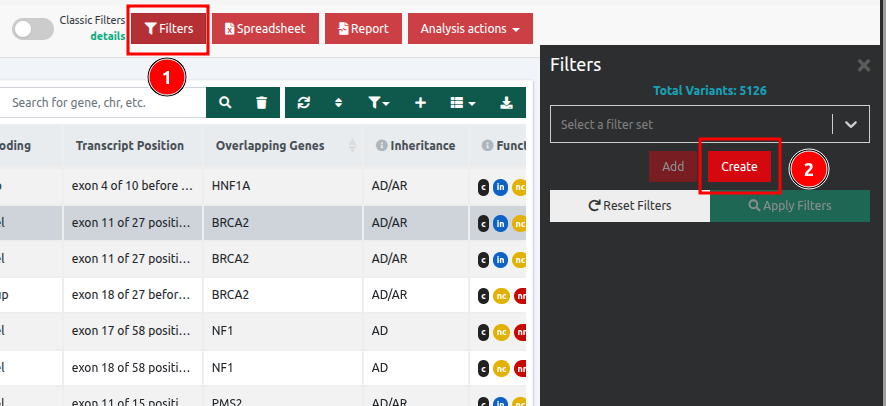
You can also create a dynamic filter to filter variants based on these values following the steps below:


How are the phenotypes matched with the variants?
The phenotype names can come from The Human Phenotype Ontology (HPO), Online Mendelian Inheritance in Man (OMIM®) or Mondo Disease Ontology (MONDO), depending on the user selection. This will affect the way the gene-phenotype associations are retrieved since we will use different sources for each phenotype term:
- HPO: in this case the HPO database is used to get the phenotype (HPO term) - gene associations.
- MONDO: the Monarch data source is used here to get the disease (MONDO term) - gene associations.
- OMIM®: the disease (OMIM®) - gene associations are retrieved directly from the OMIM® database.
