VarSome Clinical customers
If you are a VarSome Clinical customer and your assay is not yet available in VarSome Clinical, we can add your assay if it is an assay we can support.
If you need us to add a new assay, the first question is whether this is a standard assay or if it requires special handling. Examples of special handling include but are not limited to assays with Unique Molecular Identifiers (UMIs), assays that are designed to produce mixed RNA and DNA sequencing data, assays targeting specific classes of variants such as certain large deletions in genes like CFTR and various other special cases. If you do not know if an assay requires any special treatment, please talk to your assay providers and ask them if any special bioinformatics approaches are required to handle the data the assay will be used to generate.
You can now request to add your assay of preference by filling the corresponding form which is available in the following links depending on the installation you are working from:
- https://ch.clinical.varsome.com/new-assay-request/
- https://eu.clinical.varsome.com/new-assay-request/
- https://us.clinical.varsome.com/new-assay-request/
- https://sa.clinical.varsome.com/new-assay-request/
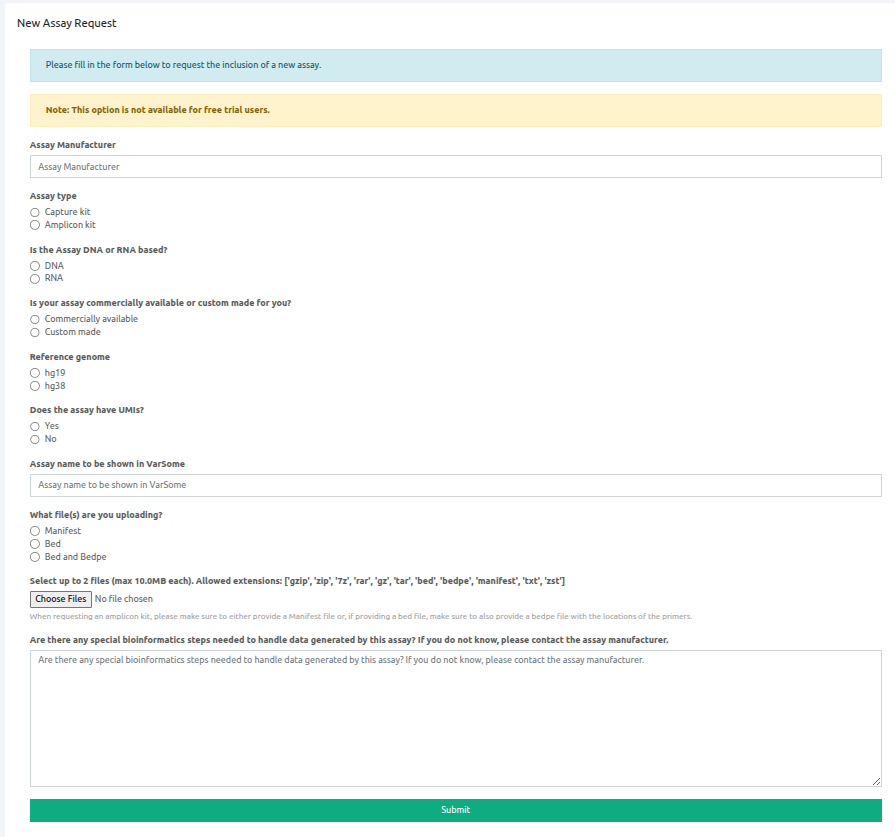
On this form you will need to fill the requested fields:
- The assay's manufacturer name
- Whether it is a capture or amplicon-based assay
- Whether it is a DNA or RNA-based assay
- Whether it is a commercially available or custom made assay
- The version of reference genome (hg19 or hg38)
- If the assay includes Unique Molecular Identifiers (UMIs)
- The name of the assay to be shown on VarSome Clinical
- The type of files you are uploading (manifest, .bed or .bed and .bedpe)
- Choose the corresponding files
- Specify if any special bioinformatics approaches are required to handle the data the assay will be used to generate.
Please keep in mind that for assays targeting relatively small regions of the genome (i.e. assays that are neither whole exome nor whole genome) variant calling will be performed in targeted mode, only looking at the regions mentioned in the assay.
If you ask us to add a new assay targeting mitochondria on hg19/GRCh37 reference genome, please use the rCRS “MT” coordinates in the accompanying BED file.
Prospects evaluating VarSome Clinical during the free trial period
Unfortunately, we cannot add assays for free trial users. However, this should not be a problem. If your assay is not listed, you can choose the "Generic capture kit", for the case of capture-based libraries.
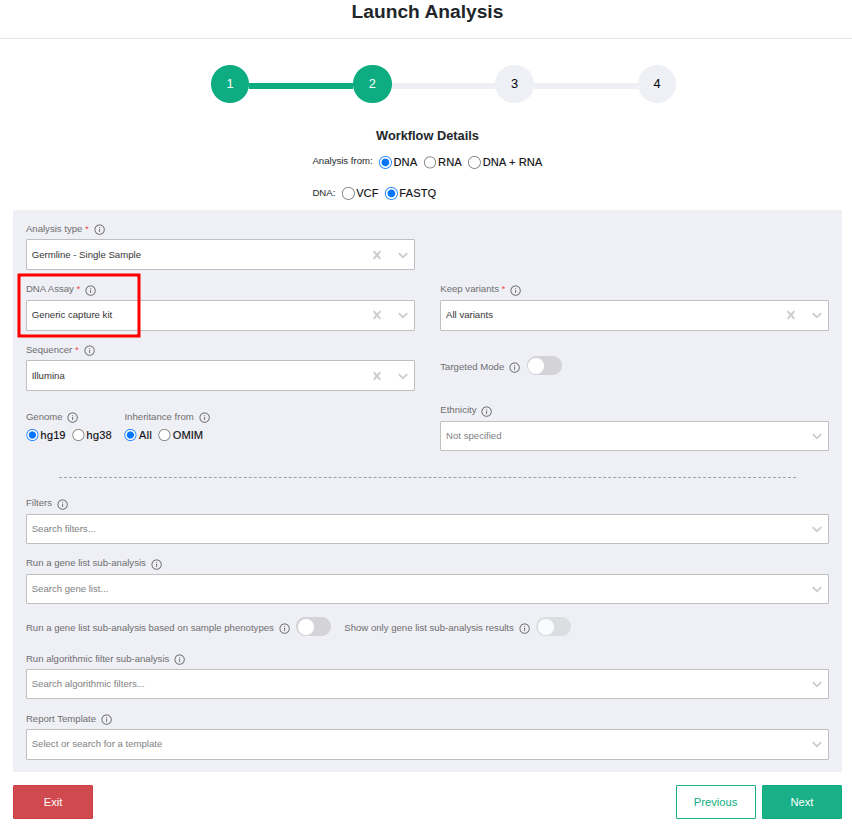
Please note that the samples analyzed using one of the "Generic" kits cannot be used for CNV sub-analyses.
As long as you choose the right chemistry type as described above, the results will be the same as if we had added your assay. The only differences will be that we will not give you statistics about the percentage of reads that fall on target and we will not run the variant calling in targeted mode (since we do not know the targets) so you may see variants that fall outside your regions of interest. However, the results will be correct and reliable.
The correct choice of the chemistry type is important because it affects:
- The way we handle possible PCR duplicates and
- The default thresholds set for minimum coverage needed to call a variant. For samples sequenced using an amplicon kit, since such kits tend to have very high coverage, the minimum threshold coverage is higher than for the capture kits.
Additionally, it is important to use the right reference genome for the assay defined on the main analysis, in order for the CNV analysis to be launched successfully. More specifically, if you run your main analysis with an assay that does not exist on our system for the reference genome of your choice, the analysis will be run in un-targeted mode. This means that the variant calling will proceed without issues, but it will include regions outside the assay's specific areas. However, when you perform a CNV analysis, the genomic regions defined by the assay are used to identify CNVs. If you attempt to run a CNV analysis using a specific assay that does not exist on our system under the reference genome chosen for the main analysis, then the following error will appear since the assay regions are not defined for that reference genome. In this scenario, you will need to re-run the main analysis using the reference genome of the assay that exists on our system, in order to be able to proceed the CNV analysis successfully.

The assay's information will be used to calculate alignment statistics and coverage of the targeted regions. The statistics are used to generate the QC report.
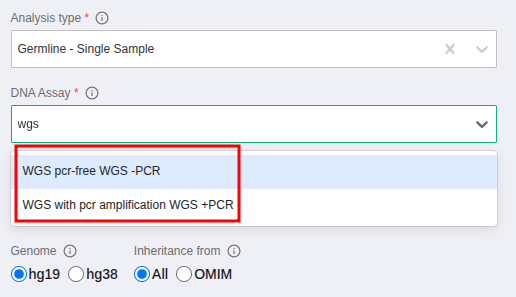
If you want to run a whole genome analysis you need to select the 'WGS' option when launching the analysis, as shown on the pic below. This option will guarantee the sample will be available for subsequent CNV analysis, if you wish to perform it.
⚠️ Please note that as VarSome Clinical can process any kind of NGS data, the list of Assay is very long. To eliminate the need to scroll through the long list of Assay whenever you are launching an analysis, you or your supervisor can simply mark certain assays as favorite. Remember to check your settings for favorite assays.
If you finally decide to join us and become a VarSome Clinical customer, we will be happy to add your assay to our platform, as described above.