Is the token valid for any of the VarSome API environments?
NO - The token is limited to one of the three available VarSome API environments.
- Live: https://api.varsome.com/
- Staging: https://staging-api.varsome.com/
- Stable: https://stable-api.varsome.com/
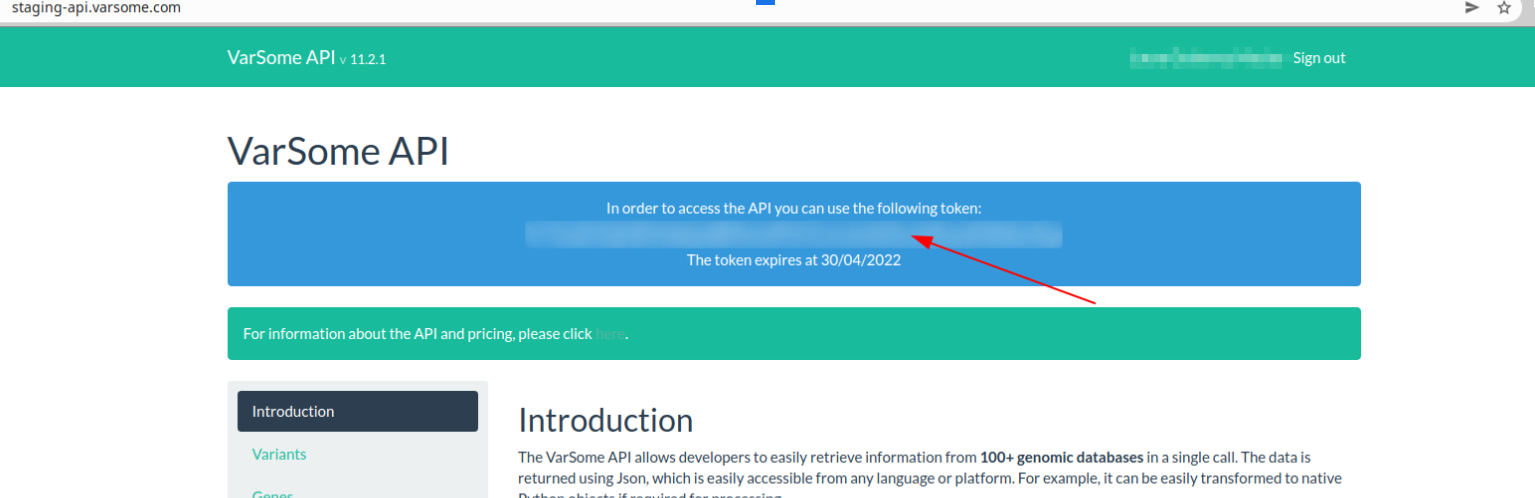
Please, make sure you are using the correct URL for the token you have been provided with. Remember that your token is available when you log in the main page e.g.:
Is there a client for the VarSome API?
YES - There is a basic API client implementation written in Python (https://github.com/saphetor/varsome-api-client-python) for https://api.varsome.com/. This client is still in beta but it is a good starting point for testing the API performance.
If you are using the client against the Staging or Stable environment, you will need to add an additional parameter (-u) to your command e.g.:
varsome_api_run.py -k $key -u 'https://staging-api.varsome.com' -q 'chr17-81195210-T-A' -p add-all-data=1
I was given a test token to evaluate the VarSome API, does this token have any limitations?
YES - Tokens are limited to the current month, meaning that if you reach the limit during the first week of the month, you won’t be able to continue testing until the next month. The limitations will depend on the environment:
- Staging API test token: limited to 10 GB per month.
- Stable or Live API test token: limited to chromosome 17 queries with 100,000 variants per month and 500 non-variant (e.g. genes) queries.
Are batch requests allowed in the VarSome API?
YES - Batch requests are supported for SNPs and small indel variants. Batch requests are not supported for CNVs at the moment.
How many variants can I query on each batch?
The number of variants in each request will be limited to the API environment.
- Staging API: this is a testing environment and the batches are limited to 50 variants maximum.
- Stable or Live API: To ensure reliable and efficient performance of VarSome API for all users, batch queries now support up to 200 variants for germline data and up to 50 variants for somatic data per request. Users can still send multiple requests. The optimal number of variants depend on different conditions. The more variants you have in each batch the bigger load you will get on the network. We recommend you experiment with the number and see what works most efficiently for your connection.
Can I just retrieve VarSome's germline classification without additional information?
NO - We always return all information that was used to calculate the germline classification alongside the classification verdict (e.g. population frequencies, pathogenicity scores, clinical evidence…). If you want to get the germline classification you need to use the add-ACMG-annotation parameter. For example, a valid query using this parameter would be:
https://api.varsome.com/lookup/15-73027478-T-C?add-ACMG-annotation=1