VarSome Clinical can annotate short tandem repeats (STR) VCF files produced by Oxford Nanopore and Illumina (Dragen). The VCFs uploaded to annotate STRs must meet the following requirements:
- Are compliant with the VCF standard.
- The number of repeats is shown in the ALT field as < STRn > where n is the number of repeats for ONT, Dragen (ExpansionHunter).
- The INFO field contains the repeat unit in the following format:
RU=GCC; SVTYPE=STR
Useful information
- Minimum pathogenic repeat counts and maximum normal repeat counts are now retrieved from the gnomAD STR dataset.
- The tandem repeat genomic region (POS–END) and Repeat Unit (RU) fields are compared against the reference region and primary disease-associated repeat unit defined in the gnomAD STR dataset. Tandem repeats that fall within the reference region and share the same repeat unit as the locus of interest will display the corresponding gnomAD STR data. Additionally, the matching also includes adjacent repeats specified in the Locus Structure field of the gnomAD STR dataset and the ExpansionHunter variant catalog, when such structures are provided.
- The gnomAD RU is additionally checked against: "Display RU" if no match is found in the RU field.
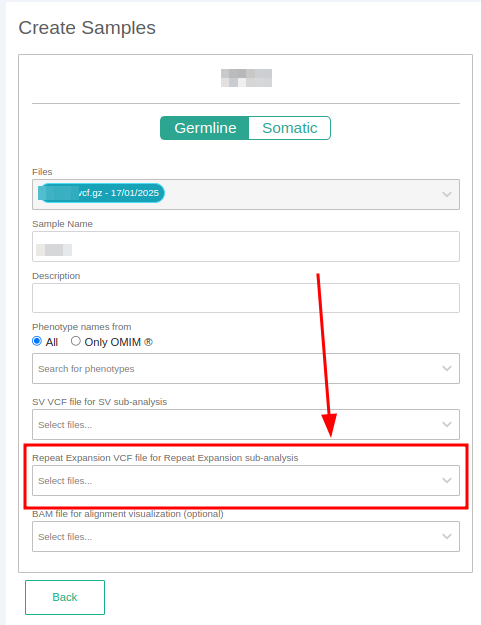
The STR analysis from VCF is launched as a sub-analysis of the main analysis. You can launch a STR annotation by:
- Adding an STR VCF file when defining your sample.

- Launching the analysis once the main analysis has finished as a “New Repeat Expansion sub-analysis” either from single or multi sample analyses.