
A case of a "problematic" variant: rs55960271. This variant is present as heterozygous in a patient and it has been annotated as pathogenic according to the ACMG Guidelines, along with ClinVar entries and known literature. However, it has been shown >700x heterozygous and 6x present as homozygous in GnomAD.
The first thing we noticed is that the variant's rsID refers to two separate variants: one is a C>A and the other a C>T. The C>A is synonymous while the C>T, the variant mentioned, is introducing a stop codon. It is therefore not impossible that some of the publications may have mistaken the variant, especially if they were performed on automatically curated data and that might be affecting the frequencies shown in the various databases. In addition, while GnomAD tries to compile data from healthy individuals, there are bound to be mistakes. Both because an individual might not be aware of any disease they may have and because the disease might simply not have manifested itself yet. Therefore, frequency alone, while a strong indicator, cannot be used as an absolute measure of a variant's pathogenicity. This C>T change will introduce an in-frame TGA stop codon near the beginning of the transcript's last exon. Since this is the last exon, the transcript with the in-frame stop codon will not be subjected to nonsense-mediated decay, and will therefore likely be translated, leading to a truncated protein. The fact that the variant appears so often in GnomAD would suggest that the truncated protein is still capable of performing its main functions. Indeed, looking closer, we noted that the variant falls outside any annotated protein domains. As you can see in VarSome, the variant (shown with the arrow) is just beyond the end of the CBS2 domain the protein carries:
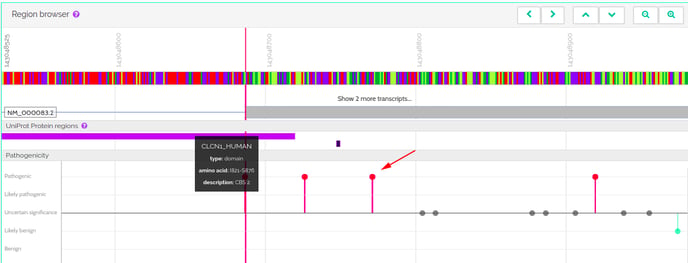
It is therefore plausible that the variant, although truncating, may still allow the resulting protein to function correctly. This variant is classified as pathogenic because it triggers rules PVS1 (null variant in a gene where LOF is a known mechanism of disease), PP3 (multiple computational predictors classify it as pathogenic) and PP5 (annotated as pathogenic in ClinVar). Since loss of function in gene CLCN1 is indeed known to cause disease, the PVS1 rule was correctly applied. There is no way of knowing that this particular variant doesn't affect the gene's protein product's function without a functional study to investigate its effects. While it is reasonable to infer that it may well not be pathogenic based on its frequency in GnomAD and the fact that the variant doesn't affect known functional domains, this doesn't change the fact that it is a null variant in a gene where loss of function is a known mechanism of disease and therefore the PVS1 rule was correctly applied. The ACMG guidelines state that if a variant fulfills the criteria for PVS1 and any two or more of the Supporting rules (PP1-PP5), then it should be classified as pathogenic. And this is precisely what has happened here. This example serves to illustrate why automated approaches can only go so far and a human brain is still needed. The ACMG guidelines [1], which we use to compute our verdict on a variant's pathogenicity [2], are guidelines. They aren't intended to be used as absolute rules. This is also why we have implemented our Germline Variant Classification algorithm in a way that allows you, the user, to activate or deactivate rules we did not apply based on knowledge that you have. We cannot decide to automatically rule out variants that look pathogenic based on their frequency alone.