Uploading files
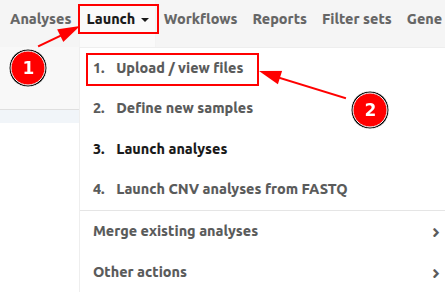
To upload files to VarSome Clinical, click on Launch > 1. Upload / View files as shown in the following picture.
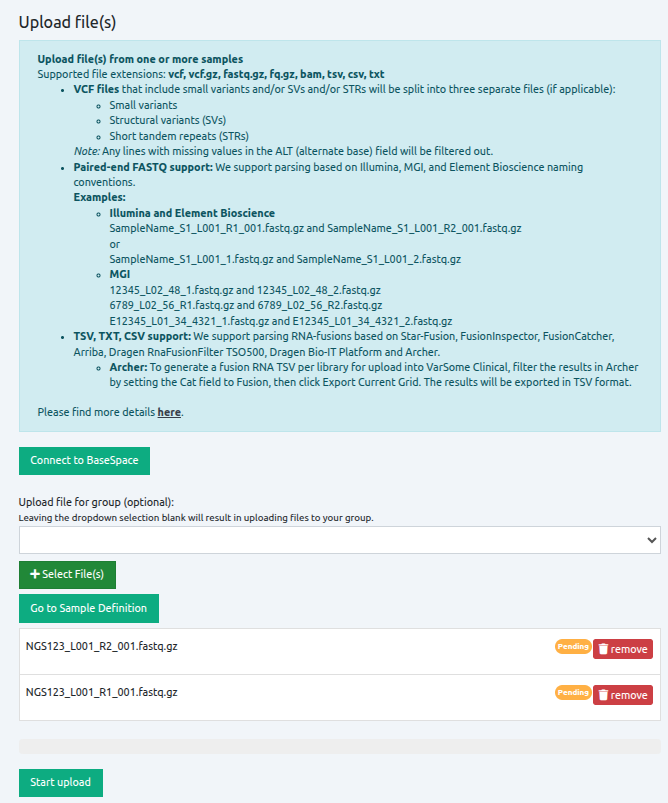
To open the file browser, click on "Select File(s)", and select all files that you want to upload. Files do not need to be from the same sample or in the same format. Once all files have been selected, the file names are displayed under the green icon "Select File(s)". To upload the files, click on "Start Upload".
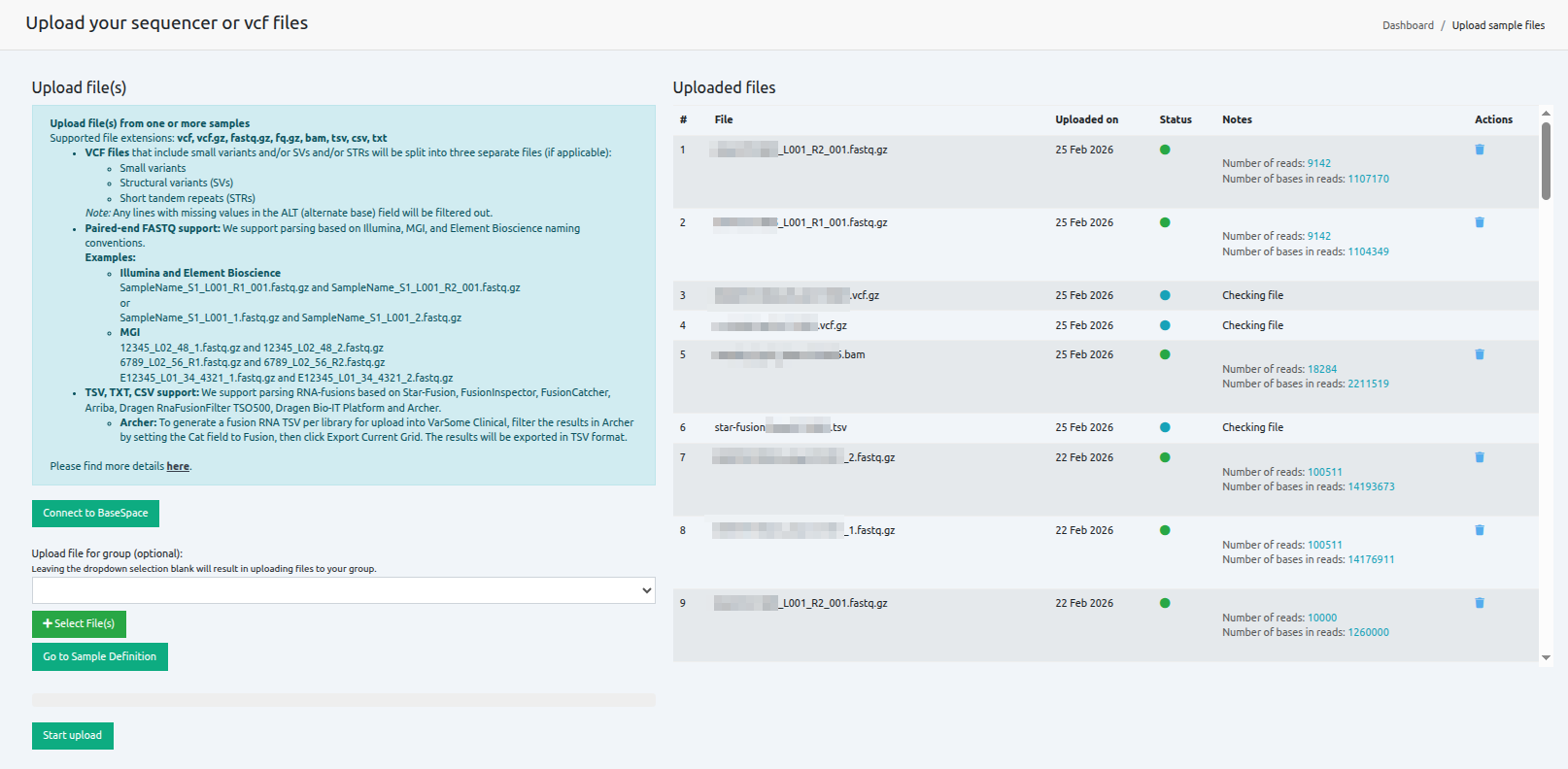
View your uploaded files
Files are uploaded and analyzed, and the number of reads and bases in reads is calculated and displayed for each file. Files can be deleted before or after upload. Files with status set to dark green can be used for a subsequent analysis.
You can organize your samples using Sample tags. For more information, please visit Sample tags.
⚠️ Please note that when uploading files, you must remain on the upload page. If you try to close the tab or browser, a pop-up will appear asking for confirmation to leave the page.
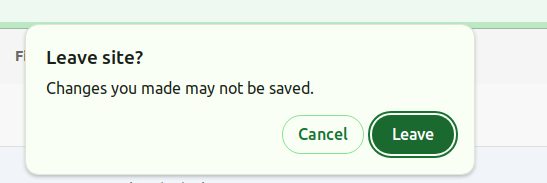
If you leave the page, the file upload process will stop. However, if the uploading reaches 100% (Uploaded) and VarSome Clinical is still validating the files on the right side, you can safely leave the page.
Additionally, if you wish to view the 'Analyses' table while the files are being uploaded, you can easily do so by opening it in a new tab.
⚠️ Please note that files that have been uploaded (free of charge) and not used for more than 30 days will be automatically deleted from VarSome Clinical.
Accepted input files
The accepted input files to run analyses on VarSome Clinical are either:
- FASTQ files only from Illumina, MGI and Element Biosciences sequencers.
- VCF files which conform to the VCF standard, regardless of sequencing platform. You can upload VCFs containing only CNVs or a mix of CNVs and other SVs. The VCFs may contain the following types of variants:
- CNVs: deletion and duplication
- Insertions
- Inversions
- Breakends
- Repeat expansions
- TSV, TXT and CSV files produced by multiple fusion detection tools from RNA-seq like Star-Fusion, FusionInspector, FusionCatcher, Arriba, Dragen RnaFusionFilter TSO500, Dragen Bio-IT Platform and Archer.
To generate a fusion RNA TSV per library for upload into VarSome Clinical using Archer, please filter the results in Archer by setting the Cat field to Fusion, and then click Export Current Grid. The filtered results will be exported in TSV format, which can then be uploaded into VarSome Clinical.
Users may also optionally upload an alignment BAM file for the VCF sample, which can be used to visualize the coverage of the variants provided in the VCF file.
Find more info in the documents:
VarSome Clinical API
The platform comes with full API, allowing you to automate each step of the data analysis process, including the data upload.