
The most common problems related to missing variants are usually twofold. First, at positions with high coverage (e.g. >3000), the Variant Caller will "down-sample" and select a random group of reads to perform the calling on. It is therefore possible that the selection doesn't accurately represent the real data and the variant might be missed. However, in cases where a good percentage (50% or more) of the reads support the variant, most likely the actual problem is coming from the noise in the region. As shown in the IGV screenshot example below, there are multiple mismatches in the region around the variant (green line).
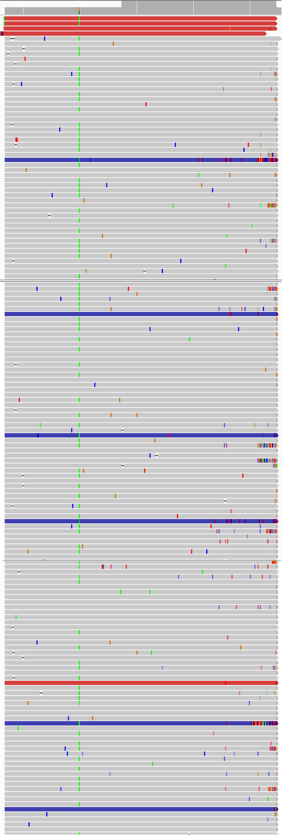
Every color indicates a mismatch and colored reads indicate paired reads whose pair was aligned to a different chromosome. Such "noisy" regions confuse the Variant Callers because when calling, they don't only look at the target position but attempt to call by region haplotype. So they use the region around the variant and build a haplotype based on the region and can miss the variant of interest. Another such example could be the presence of segmental duplications, which makes each read align almost perfectly to separate locations in the genome, leading to low alignment scores.