VarSome Clinical can annotate short tandem repeats (STR) VCF files produced by Oxford Nanopore, Illumina (Dragen), and PacBio. The VCFs uploaded to annotate STRs must meet the following requirements:
- Are compliant with the VCF standard.
- The number of repeats is shown:
- in the ALT field as < STRn > where n is the number of repeats for ONT, Dragen (ExpansionHunter), and
- in the MC field for PacBio
- The INFO field contains the repeat unit in the following format:
-
-
For ONT and Dragen (ExpansionHunter):
-
RU=GCC; SVTYPE=STR
-
- For PacBio:
TRID=NOTCH2NLC; MOTIF=GGC; MC = 6,12
Useful information
- Minimum pathogenic repeat counts and maximum normal repeat counts are now retrieved from the gnomAD STR dataset.
- The tandem repeat genomic region (POS–END) and Repeat Unit (RU) fields (for ONT and DRAGEN), or the MOTIF field (for PacBio), are compared against the reference region and primary disease-associated repeat unit defined in the gnomAD STR dataset. Tandem repeats that fall within the reference region and share the same repeat unit as the locus of interest will display the corresponding gnomAD STR data. Additionally, gnomAD matching also includes adjacent repeats specified in the Locus Structure field of the gnomAD STR dataset and the ExpansionHunter variant catalog, when such structures are provided.
- For ONT data, the gnomAD RU is additionally checked against:
Display ‘RU’ if no match is found in the RU field.
- For PacBio-derived VCFs, matching is performed against the primary disease-associated repeat unit only for this first phase.
An example of a PacBio VCF header + its corresponding variant would be the following:
##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##INFO=<ID=TRID,Number=1,Type=String,Description="Tandem repeat ID">
##INFO=<ID=END,Number=1,Type=Integer,Description="End position of the variant described in this record">
##INFO=<ID=MOTIFS,Number=.,Type=String,Description="Motifs that the tandem repeat is composed of">
##INFO=<ID=STRUC,Number=1,Type=String,Description="Structure of the region">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=AL,Number=.,Type=Integer,Description="Length of each allele">
##FORMAT=<ID=ALLR,Number=.,Type=String,Description="Length range per allele">
##FORMAT=<ID=SD,Number=.,Type=Integer,Description="Number of spanning reads supporting per allele">
##FORMAT=<ID=MC,Number=.,Type=String,Description="Motif counts per allele">
##FORMAT=<ID=MS,Number=.,Type=String,Description="Motif spans per allele">
##FORMAT=<ID=AP,Number=.,Type=Float,Description="Allele purity per allele">
##FORMAT=<ID=AM,Number=.,Type=Float,Description="Mean methylation level per allele">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT HM16212-FXN
chr6 170561906 . GGCAGCAGCAGCAACAACAACAGCAGCAGCAGCAGCAGCAGCAGCAACAGCAACAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCA GGCAGCAGCAGCAACAACAACAGCAGCAGCAGCAGCAGCAGCAGCAGCAACAGCAACAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCA,GGCAGCAGCAGCAACAACAACAGCAGCAGCAGCAGCAGCAGCAGCAGCAACAGCAACAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCA 0 . TRID=TBP;END=170562017;MOTIFS=GCA;STRUC=(GCA)n GT:AL:ALLR:SD:MC:MS:AP:AM 1/2:102,111:94-121,93-123:141,132:34,37:0(0-102),0(0-111):0.950980,0.954955:1.00,0.79
chr9 27573528 . CGGCCCCGGCCCCGGCCCC CGGCCCCGGCCCC,CGGCCCCGGCCCCGGCCCCGGCCCCGGCCCC 0 . TRID=C9ORF72;END=27573546;MOTIFS=GGCCCC;STRUC=(GGCCCC)n GT:AL:ALLR:SD:MC:MS:AP:AM 1/2:12,30:6-12,5-32:149,152:2,5:0(0-12),0(0-30):1.000000,1.000000:0.03,0.03
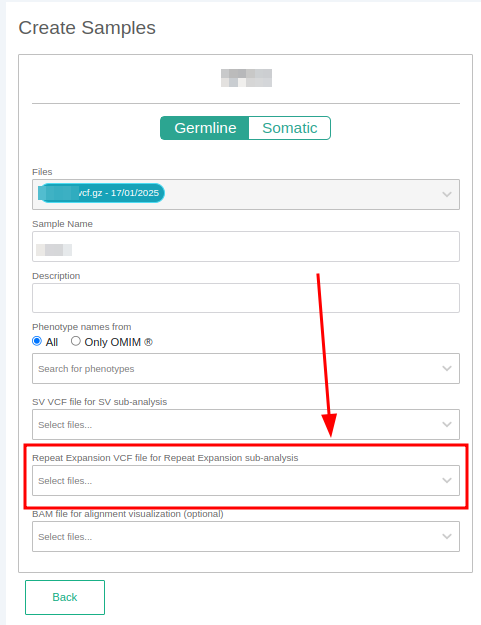
The STR analysis from VCF is launched as a sub-analysis of the main analysis. You can launch a STR annotation by:
- Adding an STR VCF file when defining your sample.

- Launching the analysis once the main analysis has finished as a “New Repeat Expansion sub-analysis” either from single or multi sample analyses.