When launching CNV/SV analyses from FASTQ, there are some common mistakes that users do that might cause issues with the launch:
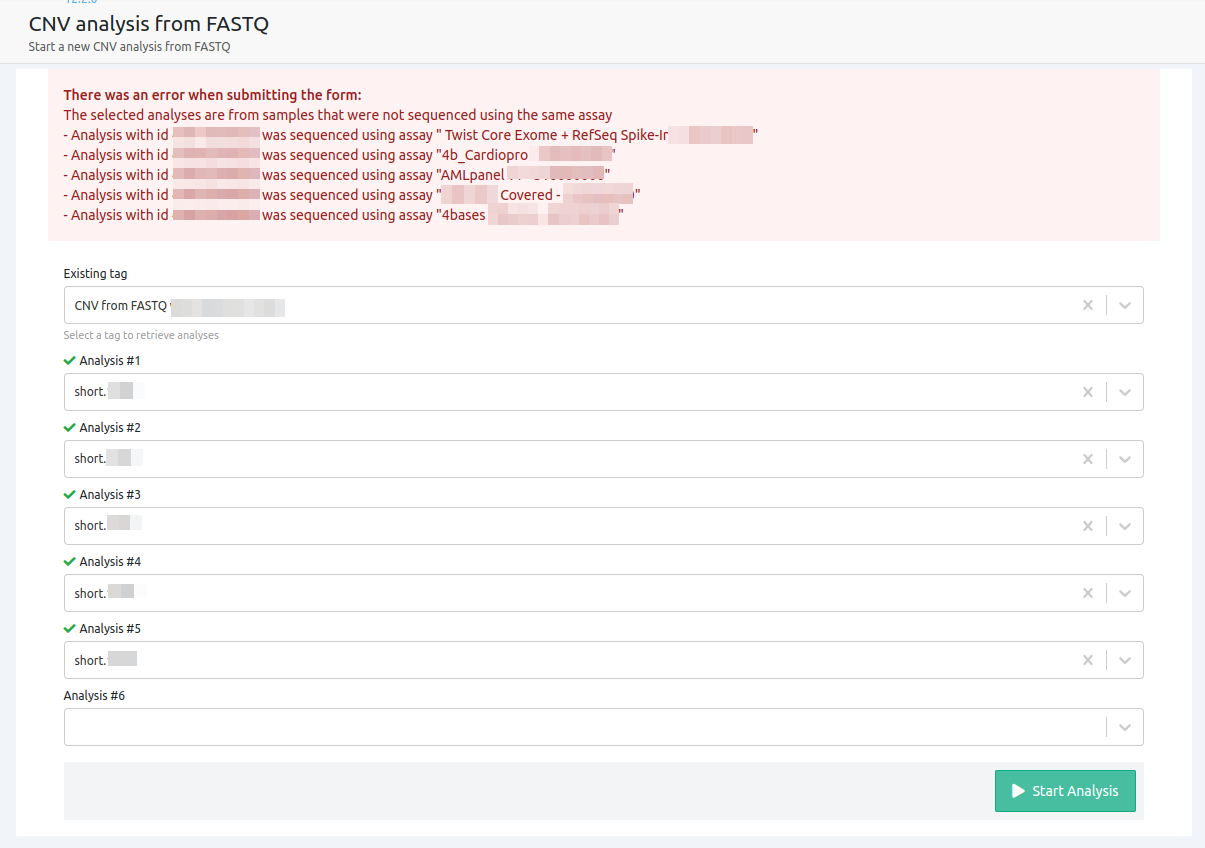
- The selected analyses are from samples that were not sequenced using the same assay
Please note that in cases where you use samples with different assays, a CNV analysis cannot run. In order for CNV analysis from FASTQ to run successfully, all the samples selected have to be run with the same assay.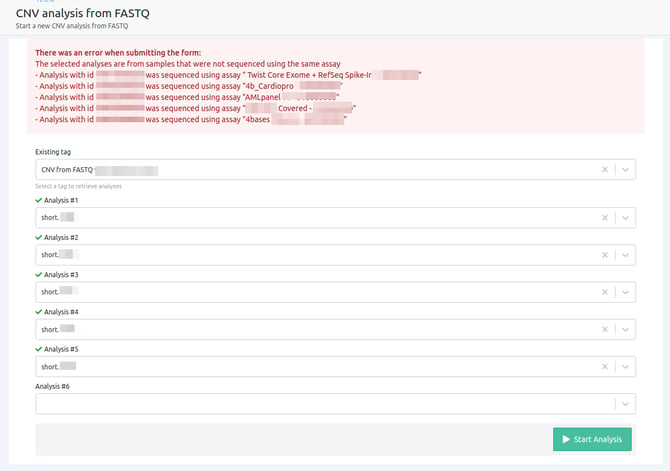
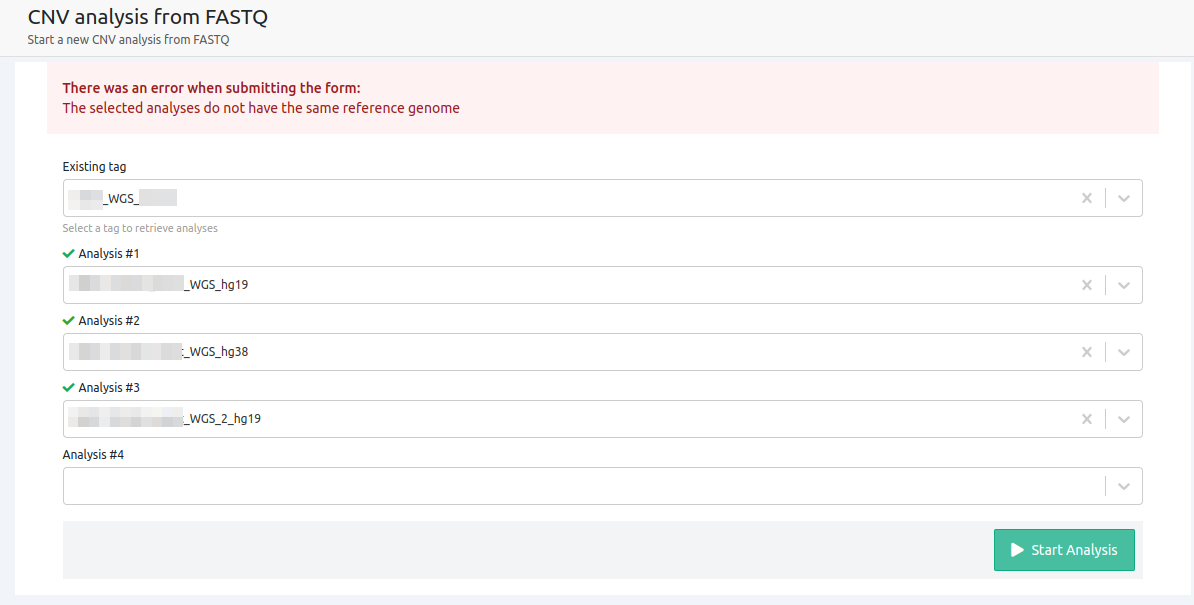
- The selected analyses do not have the same reference genome
All selected samples have to be run against the same reference genome in order to be used for CNV from FASTQ analyses.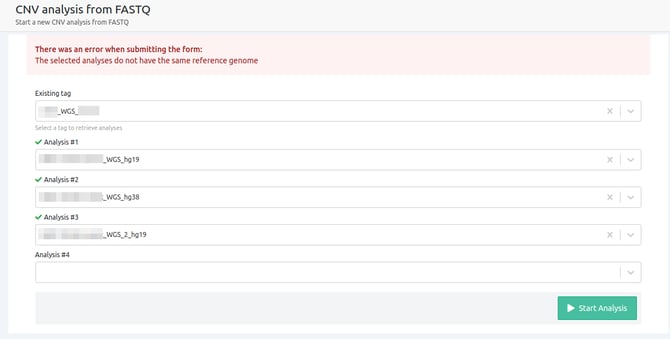
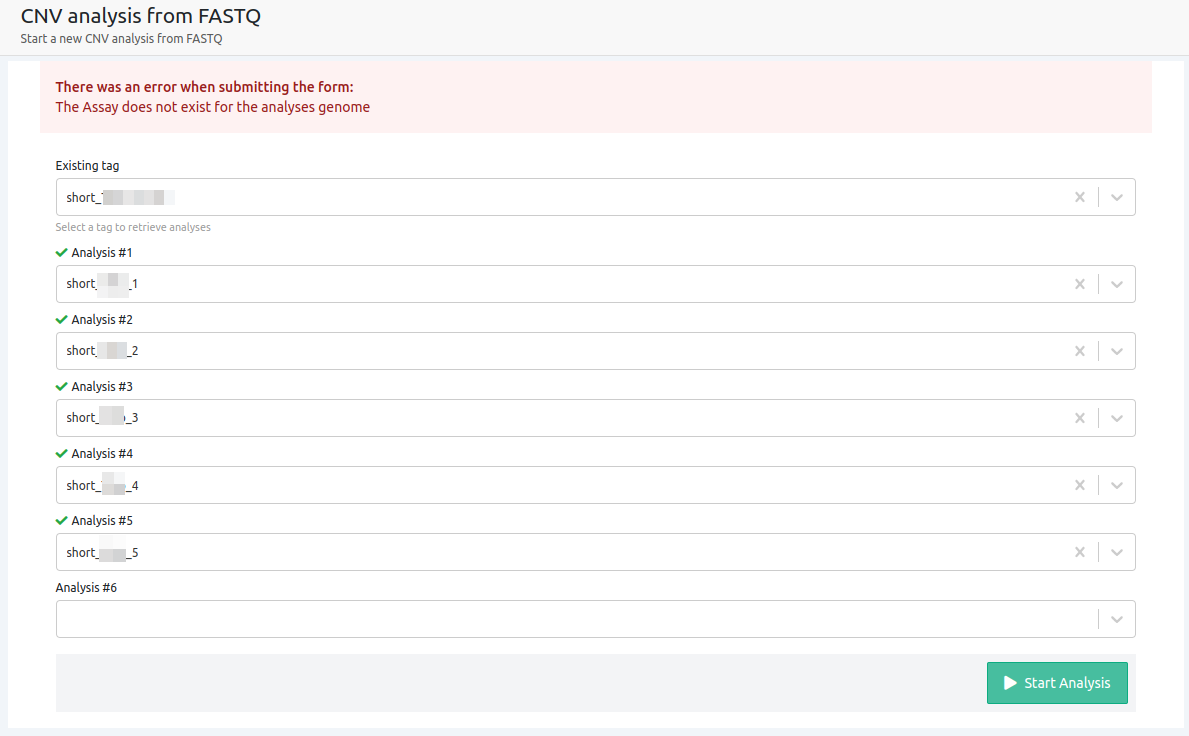
- The selected assay does not exist for the reference genome of selection
When the assay selected does not exist on our database for the reference genome of selection, the analyses will be run on un-targeted mode as there are no defined regions for the reference genome or preference to call variants from. In this case, there are no defined regions for the CNV algorithm to search for CNVs, therefore the CNV analysis cannot run.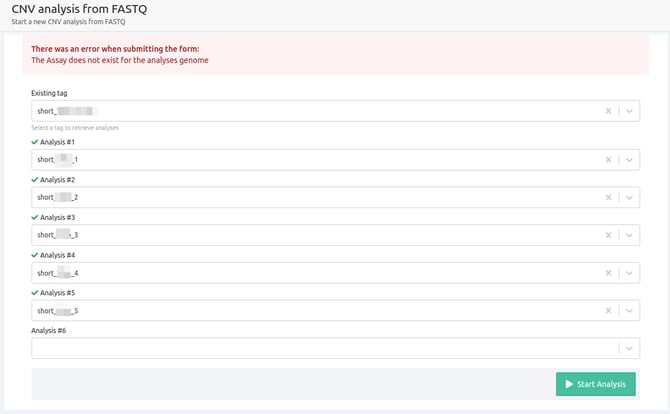
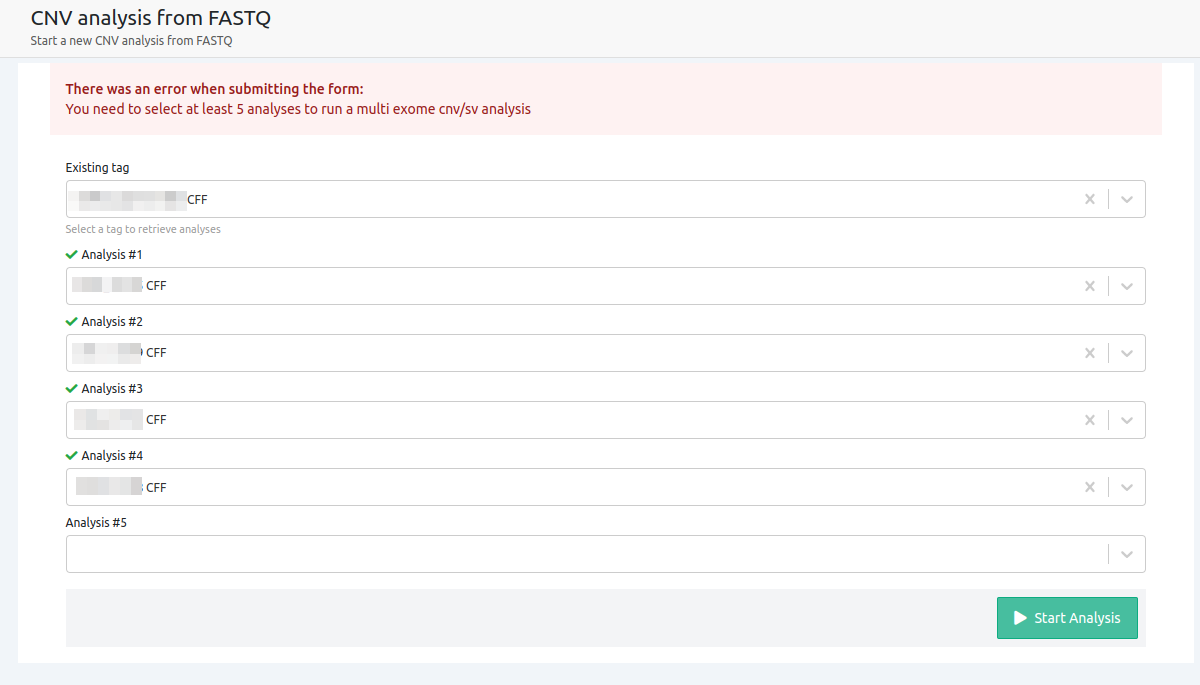
- At least 5 samples need to be selected for the analysis to be launched
To start CNV analyses from FASTQ for non WGS samples, at least 5 or more (ideally, around 10) non-WGS germline or tumor samples are needed, from the same sequencing run, as a cohort analysis.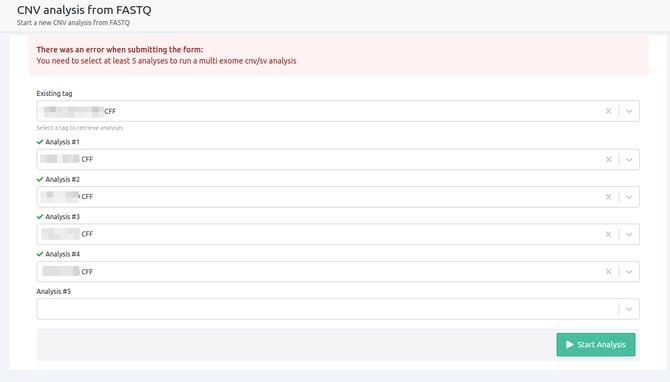
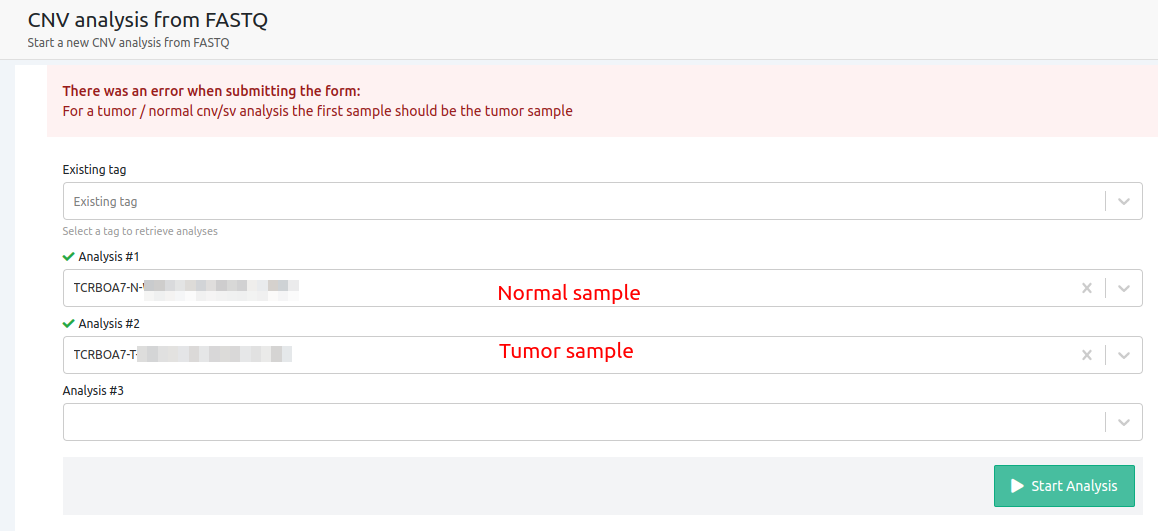
- For a tumor/normal cnv/sv analysis the first sample should be the tumor sample
In Tumor-Normal analyses, it is essential that the first sample on the selection for the CNV, will be the Tumor sample.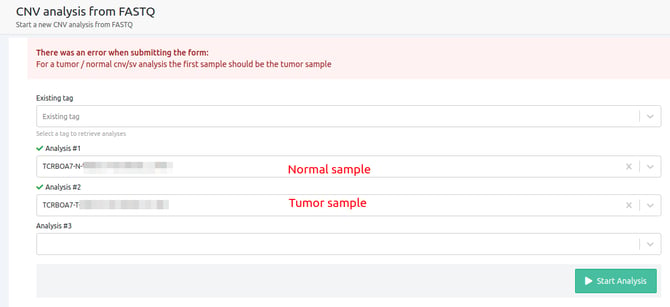
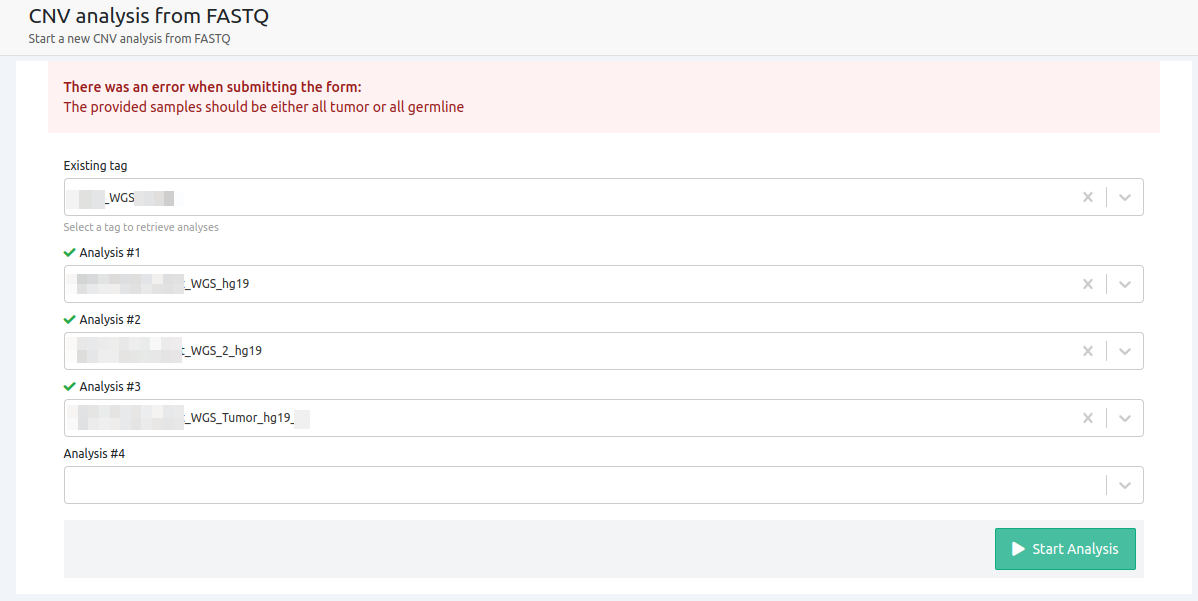
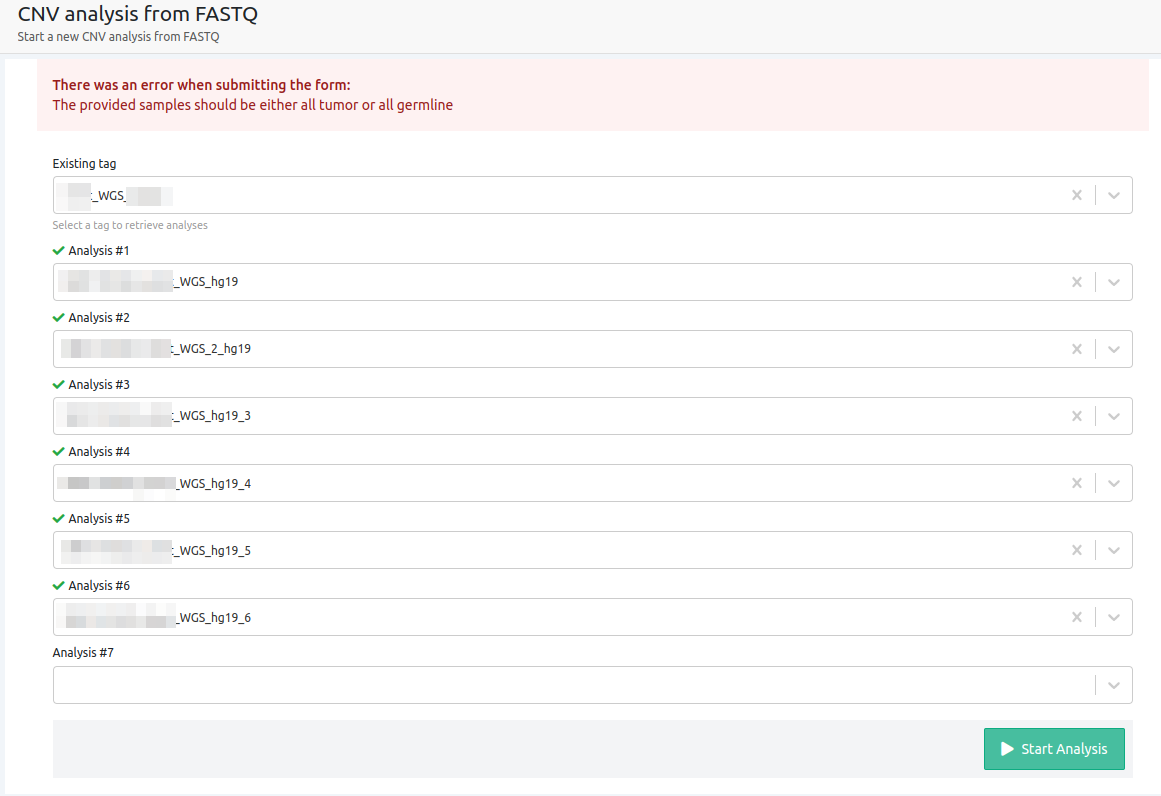
- The provided samples should be either all tumor or all germline
CNV analyses cannot run for a mixture of germline and tumor samples. This can only be performed for a Tumor-Normal analysis.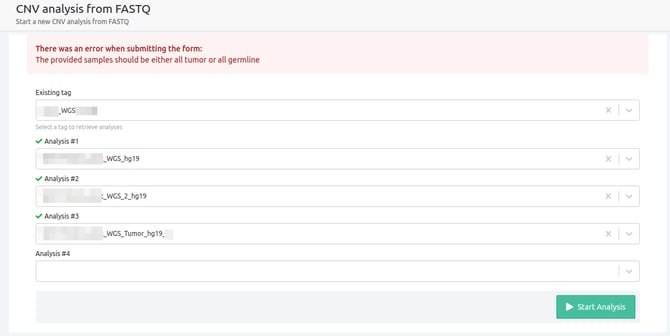
- You need to select up to 5 analyses to run a multi-sample WGS analysis
Up to 5 WGS analyses have to be selected for a multi-sample CNV from FASTQ analysis to run.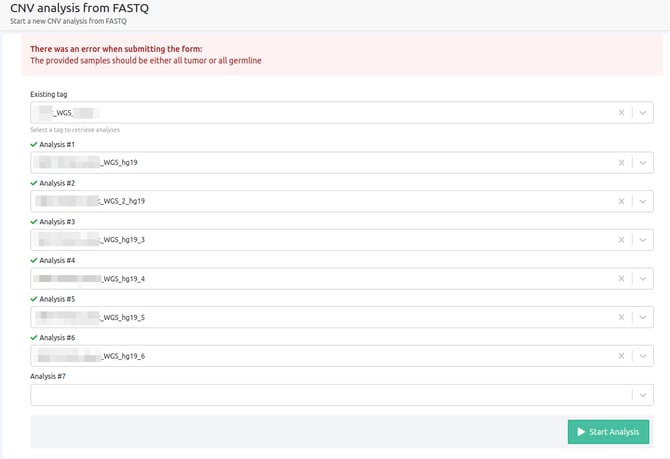
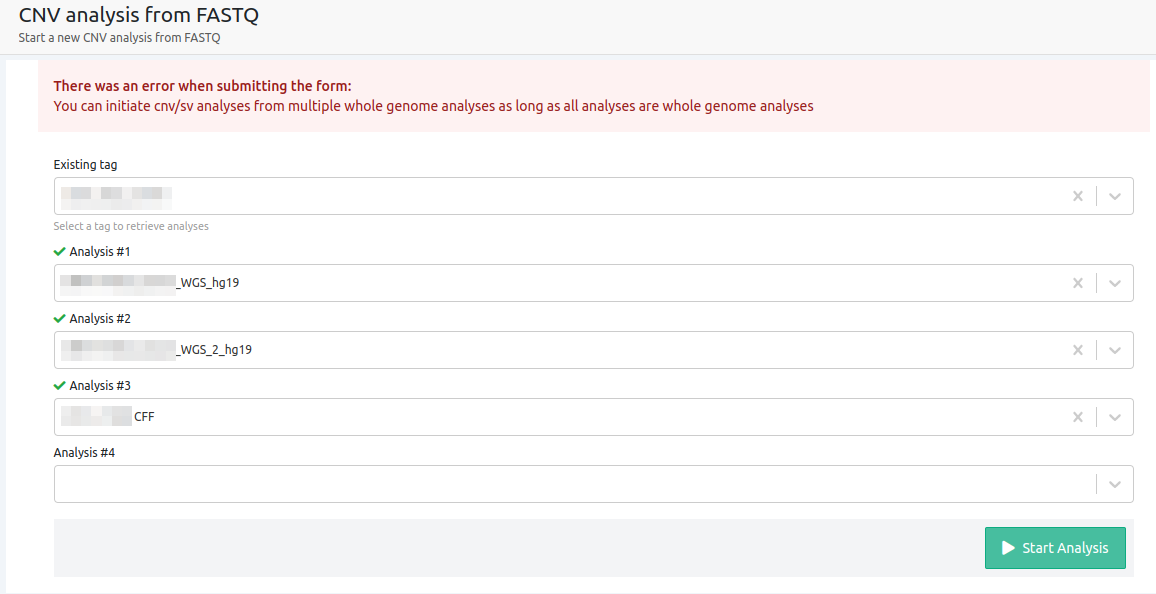
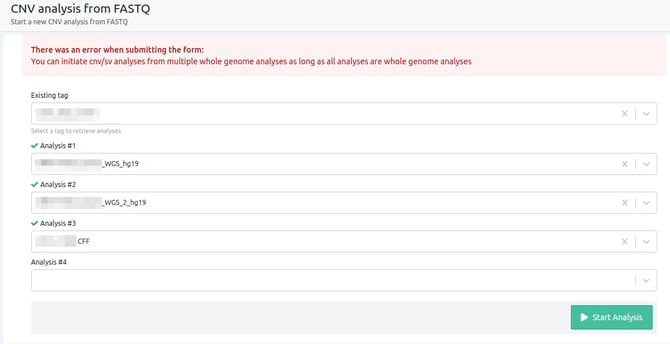
- You can initiate cnv/sv analyses from multiple whole genome analyses as long as all analyses are whole genome analyses
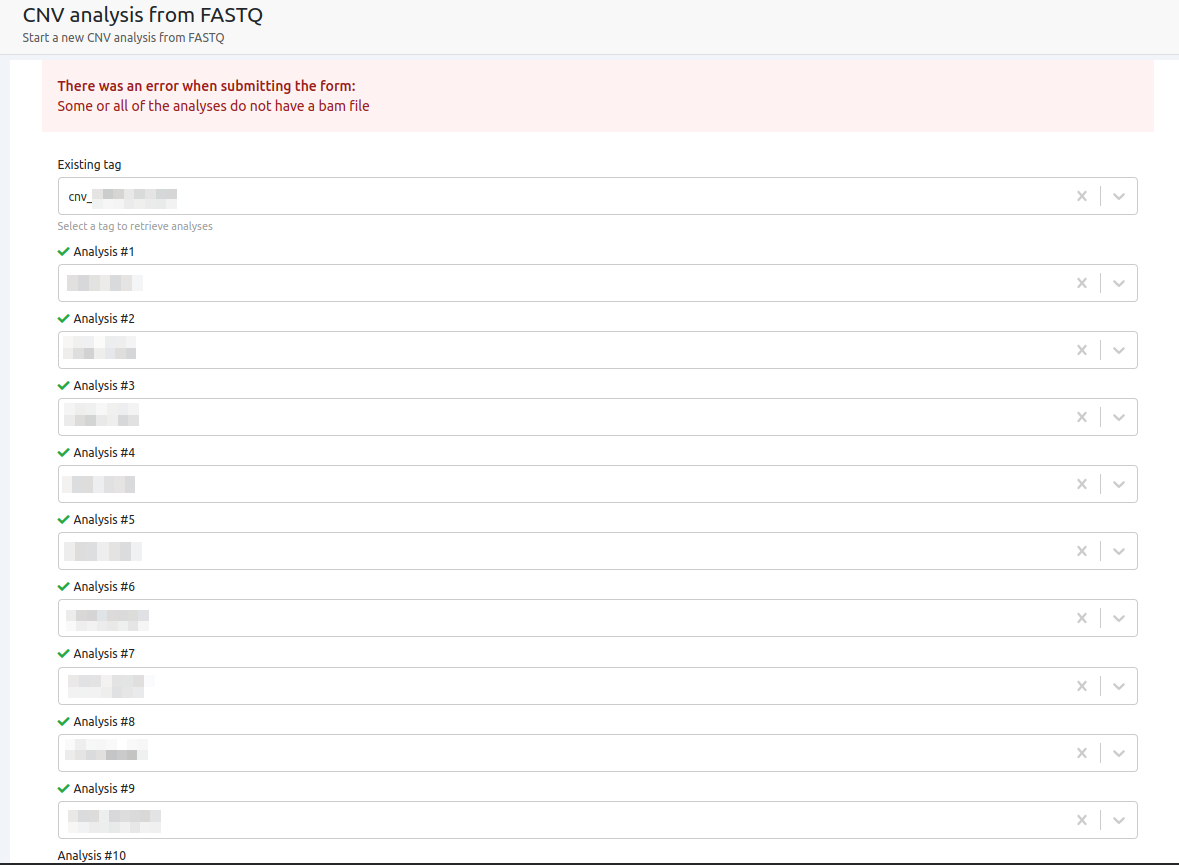
- Some or all of the analyses are lacking a BAM file
In the case where BAM files might have been deleted from one or more samples, the CNV analysis cannot run with these samples as the BAM files are essential for the CNV calling.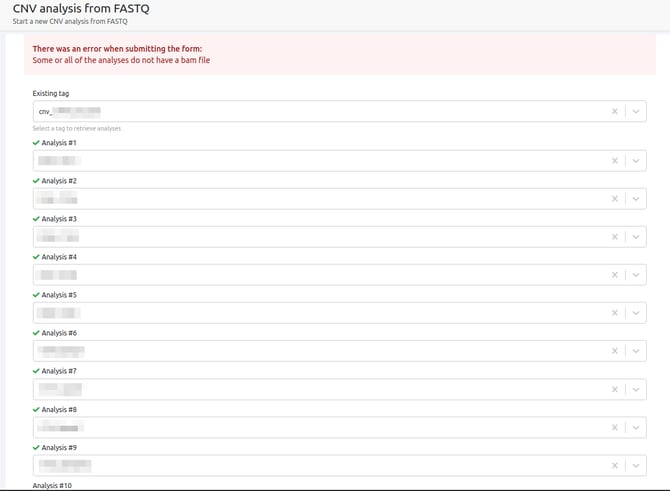
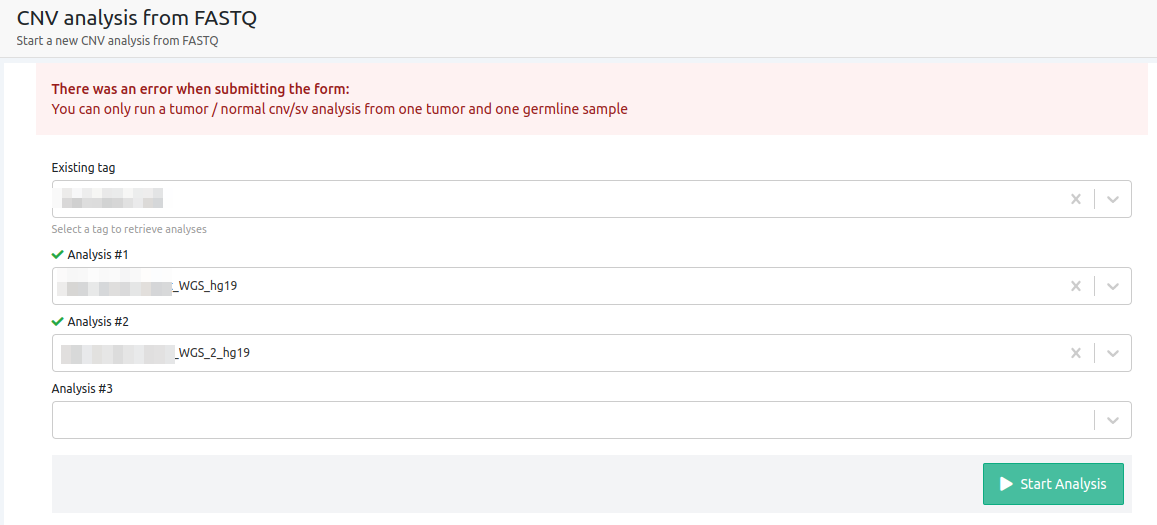
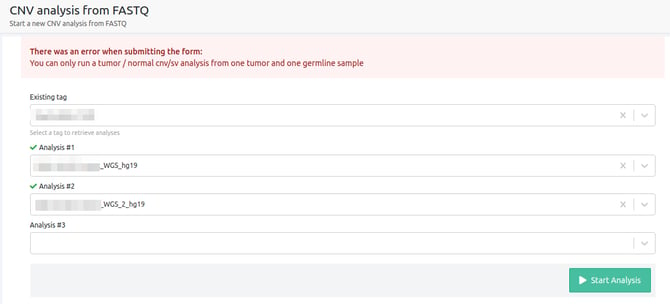
- You can only run a tumor / normal cnv/sv analysis from one tumor and one germline sample
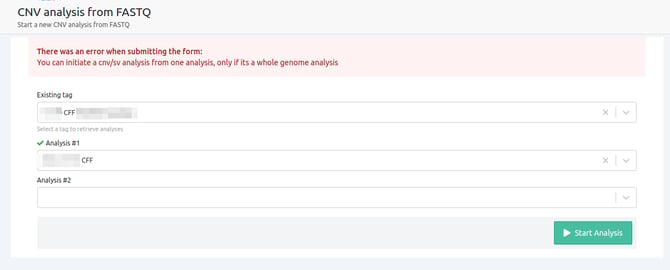
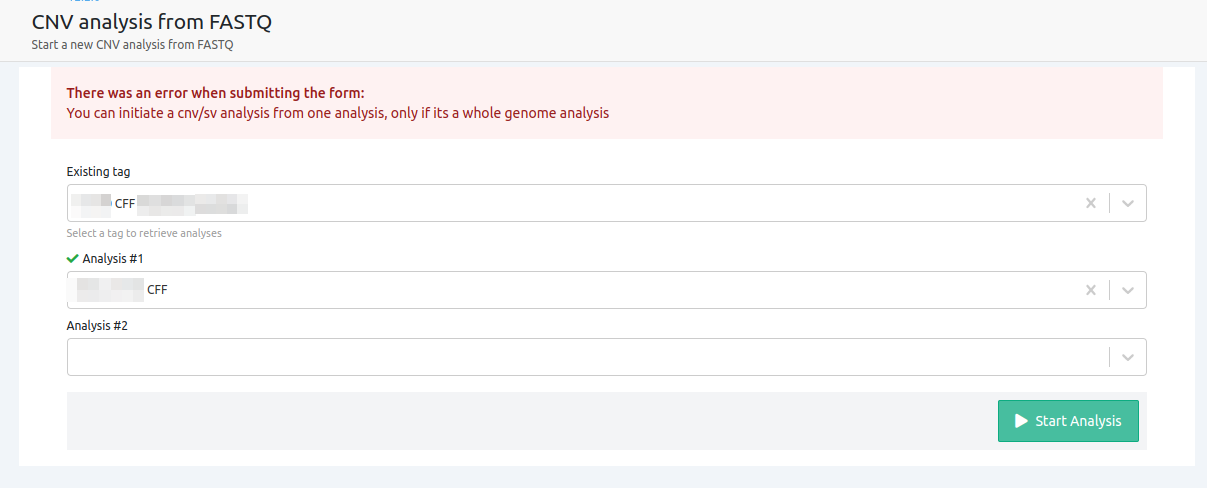
- You can initiate a cnv/sv analysis from one analysis, only if it's a whole genome analysis
The only case where a single analysis can be used to run a CNV from FASTQ is when it is a WGS analysis.