Introduction
VarSome Clinical version 13.13.1, released in December 2025, introduced a new parameter used when calling variants on single-sample analyses from capture-based (as opposed to amplicon) assays. This is a machine learning (ML) model which is leveraged in the variant calling step performed by the Sentieon DNAScope variant caller (see Sentieon's documentation).
Variant calling with DNAScope is a two-step process. In the first step, variant calling takes place using a sensitive mode with the DNAscope algorithm. The goal for this step is to identify and annotate all possible germline variants (variant candidates) that are present in the aligned read data.
In the second step, variants are filtered and genotyped using a pre-trained machine learning model. The model aims to distinguish true variants from errors caused by any part of the pipeline (e.g. library prep, sequencing, alignment etc.) as well as to provide better handling of regions inherently difficult to interpret. It considers features such as Shannon entropy, unfiltered read depth (UDP), mean base quality (NBQ), and read position distance. You can find more information in DNAScope’s publication. At this stage, variant candidates are marked with "MLrejected" if the ML model believes that the most likely genotype for a site is homozygous reference (i.e. no variant).
This change has improved the variant calling performance of VarSome Clinical, but has also caused some confusion. Users will likely have noticed that the system now calls significantly more variants. This article will explain why this change was implemented and how to deal with the higher number of variants.
Variants that are marked as “ML Rejected”, are considered “Failed/Not Genotyped” by VarSome Clinical. This means that they will be removed from the results if the analysis is run using the "Keep only variants that pass QC criteria" option. The vast majority of variants called with the ML mode that were not called without it are marked this way and are therefore marked as Failed.
Validation
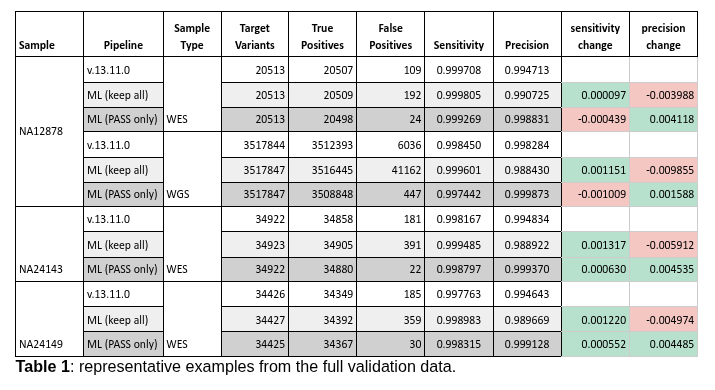
Before this change was made, we performed extensive validation to ensure that it was an improvement. Our results show that, when considering only variants marked as having passed QC (therefore excluding the variants marked as MLRejected), the ML mode greatly reduces false positives, resulting in higher precision. On the other hand, when the MLRejected variants are included, the ML mode increases sensitivity. You can see the full results of our internal validation here, and a representative example in Table 1 below.
Usage recommendation
This is therefore one of the very rare cases where a change in variant calling manages to improve both sensitivity and precision instead of there being a tradeoff between the two. In order to make the most of this feature, we recommend:
- Run all analyses with the keep all variants option

- Apply a filter to the results to keep only those variants marked as pass.

- If you haven't found the causative variants and need to look deeper, remove the filter to include all of the identified variants.
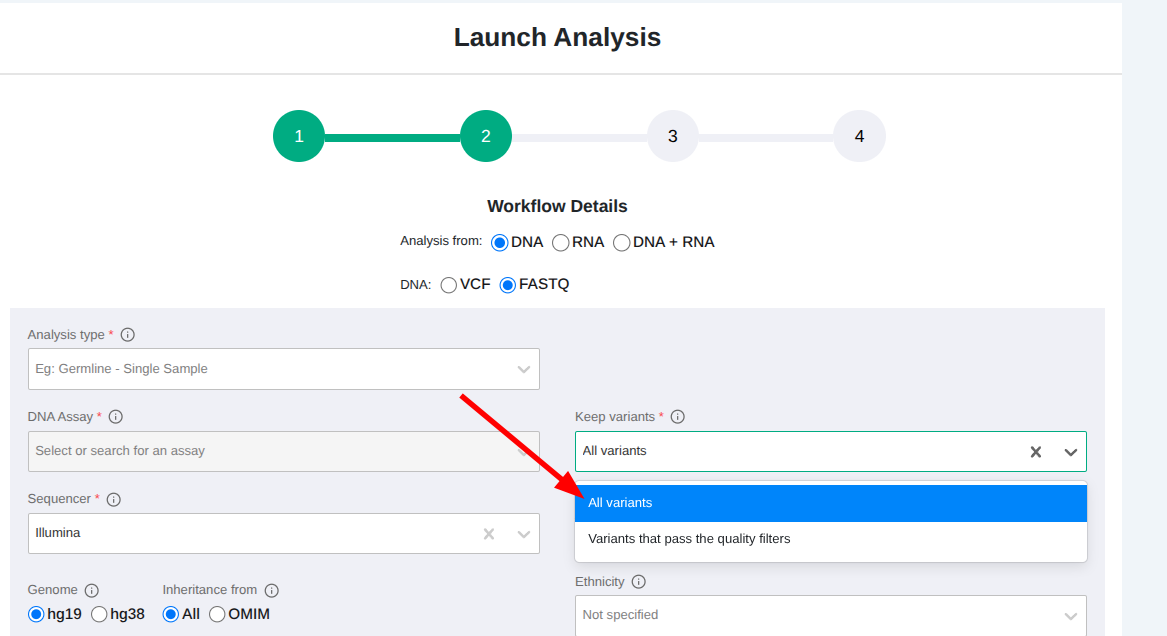
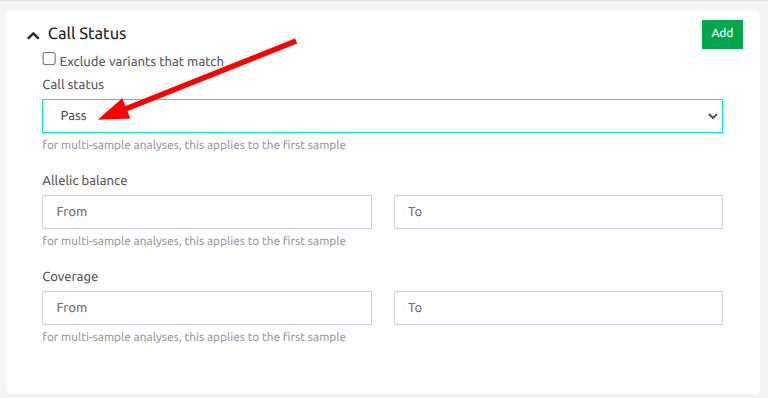
This approach is better than running with "Keep variants that pass the quality filters" as that option means that variants that fail the QC criteria are discarded and there is no way for the user to retrieve them. Using the approach above, you get the best of both worlds: you can have the increased precision of the ML model but you can still access the rejected variants if necessary.